
Abstract
Huperzine A-producing fungus Penicillium polonicum Hy4 (CCTCC No.M2010086) was isolated from Huperzia serrata (Thunb) Trev. The complete mitochondrial genome of P. polonicum is 28 192 bp in length, containing 15 protein-encoding genes, 27 tRNA genes and two rRNA genes. The whole mitogenome is high in AT content (74.40%) and low in GC content (25.60%). The mitochondrial gene order and arrangement of P. polonicum are identical to those of other Penicillium. Phylogenetic analysis based on 14 concatenated protein-encoding genes showed that P. polonicum was close to P. solitum. This study reports the complete mitogenome of P. polonicum for the first time and provides valuable information for further exploration of mitochondrial evolution.
Penicillium polonicum strain Hy4 (CCTCC No.M2010086) , isolated from Huperzia serrata in Shaoyang, Hunan, China (27.22 North, 111.50 East) was found to produce Huperzine A (Hup A) and was deposited in China Center for Type Culture Collection (CCTCC). Hup A is used to treat dementia as a reversible and selective acetylcholinesterase (AChE) inhibitor with low toxicity (Zhang & Tang Citation2006). Up to now, Hup A is mainly isolated from H. serrata which have an extremely long vegetative growth cycle and low Hup A content (Ma et al. Citation2005). Endophytic fungi are one of the most creative groups of high-value secondary metabolite producers (Venugopalan & Srivastava Citation2015).
Initial taxonomic evaluation of P. polonicum Hy4 was made based on ITS sequence (Makimura et al. Citation2001). The mitogenome was sequenced by Illumina Hiseq 2000 and assembled by Allpaths-LG (Gnerre et al. Citation2011), with two gaps filled by PCR amplification. It was a circular-mapping DNA molecule of 28 192 bp with a low GC content of 25.60% (Genbank accession number: KU530219). The mitogenome contained 15 conserved protein-encoding genes (13 728 bp, 48.69% of the mitogenome) that were discovered by searching NCBI NR database using Exonerate (Slater & Birney Citation2005; Lin et al. Citation2015). Those genes encoded ATP synthase subunits (atp6, atp8 and atp9), cytochrome oxidase subunits (cox1, cox2 and cox3), apocytochrome b (cob), NADH dehydrogenase subunits (nad1, nad2 and nad3, nad4, nad4L, nad5 and nad6) and ribosomal protein (rps3). All genes were located on one strand and apparently transcribed in one direction. Additionally, 27 tRNA genes were predicted by tRNAscan-SE (Lowe & Eddy Citation1997) and the large/small subunits ribosomal RNA genes (rns, rnl) were identified by searching against Rfam database (Burge et al. Citation2013). Similar to many filamentous fungi, P. polonicum mitochondrial tRNA genes were organized into two dense gene clusters (KGGDSWISP, TEVMMLAFLQM), which were flanked by cox3, rnl and cox1 (Sun et al. Citation2011; Eldarov et al. Citation2012; Joardar et al. Citation2012). The comparison of trn distribution in the mitogenomes showed that the trn order in Penicillium was relatively conserved in contrast to that in Aspergillus.
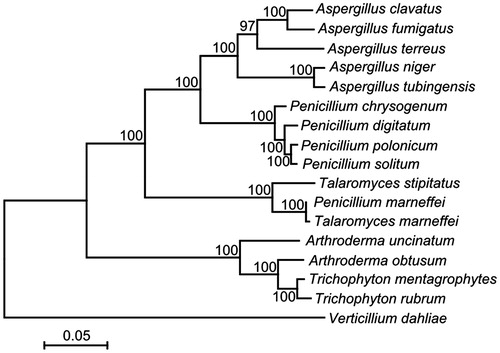
The codon frequency analysis showed that a total of 56 codons were used for transcription, with the absence of ACC, CCC, CGA, CGC, CGG, CTC, GGC and TCG. All protein-encoding sequences started with the typical ATG codon and ended with TAA as the stop codons except for nad6 and cox3 (TAG). AT-rich codons were abundant, up to 82.36%, reflecting the high AT content of the P. polonicum mitogenome. The most used codon is TTA for Leu and followed by ATA for Ile. The ratio of codons encoding hydrophobic amino acids (Met, Trp, Phe, Val, Leu, Ile, Pro, Ala) was 54.74%, reflecting the hydrophobic nature of respiratory membrane complexes. A maximum likelihood tree generated by 14 protein-coding genes from 17 fungal strains () suggested the closest relationship between P. polonicum and P. solitum. Moreover, Penicillium was closely related to Aspergillus, except Penicillium marneffei, which was positioned on a distinct branch. This observation was identical with the earlier study based on nuclear genome (van den Berg et al. Citation2008).
Figure 1. Phylogenetic relationship between P. polonicum and other 16 fungal mitogenomes. The maximum likelihood tree was generated by MEGA v6.06 (Tamura et al. Citation2013) based on 14 concatenated protein-coding genes (nad1-6, nad4L, atp6, atp8-9, cob and cox1-3). Verticillium dahliae was used as outgroup. The model for phylogenetic analysis was GTR + G+I with bootstrap value of 1000. The sequence data for these 17 fungi with complete mitochondrial genomes were used: Aspergillus clavatus (JQ354999), Aspergillus fumigatus (JQ346807), Aspergillus terreus (JQ355001), Aspergillus niger (NC_007445), Aspergillus tubingensis (NC_007597), Penicillium chrysogenum (JQ354996), Penicillium digitatum (NC_015080), Penicillium polonicum (KU530219), Penicillium solitum (NC_016187), Talaromyces stipitatus (JQ354994), Penicillium marneffei (JQ354997), Talaromyces marneffei (NC_005256), Arthroderma uncinatum (NC_012828), Arthroderma obtusum (NC_012830), Trichophyton mentagrophytes (NC_012826), Trichophyton rubrum (NC_012824), Verticillium dahliae (DQ351941).
Funding information
This study was supported by the Program of International Science & Technology Cooperation (2013DFG32060).
Acknowledgements
The authors are grateful to Runmao Lin for the kind advice in data analysis.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Related Research Data
References
- Burge SW, Daub J, Eberhardt R, Tate J, Barquist L, Nawrocki EP, Eddy SR, Gardner PP, Bateman A. 2013. Rfam 11.0: 10 years of RNA families. Nucleic Acids Res. 41: D226–D232.
- Eldarov MA, Mardanov AV, Beletsky AV, Dzhavakhiya VV, Ravin NV, Skryabin KG. 2012. Complete mitochondrial genome of compactin-producing fungus Penicillium solitum and comparative analysis of Trichocomaceae mitochondrial genomes. FEMS Microbiol Lett. 329:9–17.
- Gnerre S, Maccallum I, Przybylski D, Ribeiro FJ, Burton JN, Walker BJ, Sharpe T, Hall G, Shea TP, Sykes S, et al. 2011. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci USA. 108:1513–1518.
- Joardar V, Abrams NF, Hostetler J, Paukstelis PJ, Pakala S, Pakala SB, Zafar N, Abolude OO, Payne G, Andrianopoulos A, et al. 2012. Sequencing of mitochondrial genomes of nine Aspergillus and Penicillium species identifies mobile introns and accessory genes as main sources of genome size variability. BMC Genomics. 13:698.
- Lin R, Liu C, Shen B, Bai M, Ling J, Chen G, Mao Z, Cheng X, Xie B. 2015. Analysis of the complete mitochondrial genome of Pochonia chlamydosporia suggests a close relationship to the invertebrate-pathogenic fungi in Hypocreales. BMC Microbiol. 15:5.
- Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25:955–964.
- Ma X, Tan C, Zhu D, Gang DR. 2005. Is there a better source of huperzine A than Huperzia serrata? Huperzine A content of Huperziaceae species in China. J Agric Food Chem. 53:1393–1398.
- Makimura K, Hanazawa R, Takatori K, Tamura Y, Fujisaki R, Nishiyama Y, Abe S, Uchida K, Kawamura Y, Ezaki T, et al. 2001. Fungal flora on board the Mir-Space Station, identification by morphological features and ribosomal DNA sequences. Microbiol Immunol. 45:357–363.
- Slater GS, Birney E. 2005. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics. 6:31.
- Sun X, Li H, Yu D. 2011. Complete mitochondrial genome sequence of the phytopathogenic fungus Penicillium digitatum and comparative analysis of closely related species. FEMS Microbiol Lett. 323:29–34.
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol. 30:2725–2729.
- Van Den Berg MA, Albang R, Albermann K, Badger JH, Daran JM, Driessen AJ, Garcia-Estrada C, Fedorova ND, Harris DM, Heijne WH, et al. 2008. Genome sequencing and analysis of the filamentous fungus Penicillium chrysogenum. Nat Biotechnol. 26:1161–1168.
- Venugopalan A, Srivastava S. 2015. Endophytes as in vitro production platforms of high value plant secondary metabolites. Biotechnol Adv. 33:873–887.
- Zhang HY, Tang XC. 2006. Neuroprotective effects of huperzine A: new therapeutic targets for neurodegenerative disease. Trends Pharmacol Sci. 27:619–625.