
Abstract
The complete mitochondrial genome of the green mirid, Creontiades dilutus, a significant pest of cotton in Australia, comprises 15,864 bp and has a GC content of 22.3%. The layout of the 13 mitochondrial protein-coding genes follows the ancestral insect arrangement, and 22 tRNA’s were detected as well as the small and large rRNA’s. Phylogenetic analysis of available mirid mitogenomes places Creontiades closer to Adelphocoris than the other four genera.
The green mirid (Creontiades dilutus) is endemic to Australia and is primarily associated with the arid interior of the continent (Hereward et al. Citation2013a), although it has also become a major pest of several crops. It is regarded as a generalist species and has been associated with over 97 different plant species, but is most abundant on two native plant species, Cullen australasicum and Cullen cinereum (Hereward & Walter Citation2012). Population genetic analysis indicates that C. dilutus is a single species gene-pool across the many plants it uses, and is not a suite of cryptic species (Hereward et al. Citation2013a, Citation2013b). Sequencing of a fragment of the Cytochrome oxidase I gene has revealed very low genetic diversity at this locus (Coleman et al. Citation2008; Hereward et al. Citation2013a), with the majority of individuals being represented by a single haplotype. To provide additional mitochondrial markers and a resource for future genomic investigations into this species, I sequenced and assembled the complete mitochondrial genome using Illumina sequencing technology.
DNA was extracted from a single individual collected from Goodenia cycloptera (Goodeniaceae) in inland Australia (Lat. 26°15'56.70″S, Long. 136°11'58.86″E), the whole insect was used for DNA extraction but representative samples are stored at the University of Queensland and are available on request. Sequencing was performed by Novogene (Beijing, China) resulting in 70 × 106 paired-end 125 bp reads. The mitogenome was assembled in Geneious v9.0.4 (http://www.geneious.com, Kearse et al. Citation2012) by first mapping reads to available mirid mitochondrial genomes, de-novo assembly of mitochondrial reads and contig extension by iterative mapping until the full circularised genome was obtained. Annotation was achieved initially using the MITOS webserver (Bernt et al. Citation2013). These annotations were manually checked against other miridae mitogenomes obtained from GenBank and by mapping transcriptome reads to double check gene ends by the presence of reads containing the poly-A tail. All 13 protein-coding genes were present as well as 22 tRNA’s and the small and large rRNA’s. The gene layout follows the pattern common to most insect mitochondrial genomes (the insect ancestral arrangement) and all tRNA’s produced a cloverleaf secondary structure. The control region of C. dilutus was 1242 bp, and there is a small (122 bp) intergenic gap between Nad6 and CytB.
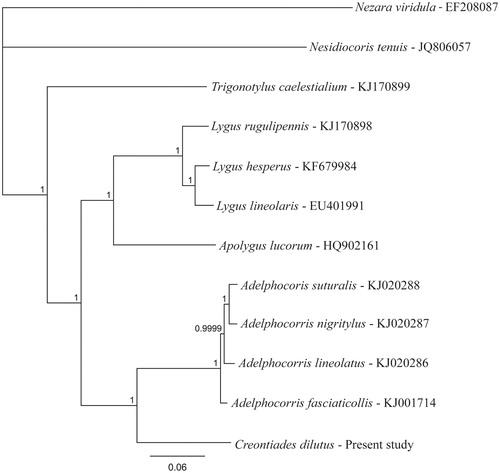
All complete mirid mitochondrial genomes were downloaded from the nt database of GenBank, and a phylogenetic tree of the core gene section was produced using MrBayes (Huelsenbeck & Ronquist Citation2001) with Nezara viridula (GenBank accession no. EF208087, Hua et al. Citation2008) as the outgroup (). This analysis placed Creontiades closer to Adelphocoris than the other four genera that have complete mitochondrial genomes. However, there is still considerable genetic distance between these genera (15.7% sequence difference between C. dilutus and A. fasciaticollis). The mitochondrial genome of C. dilutus has been deposited at GenBank under accession no. KU753807.
Figure 1. Phylogenetic tree produced using Bayesian estimation (MrBayes Inc., New York, NY) on the core gene encoding region of all available mirid mitochondrial genomes with Nezara viridula (Pentatomidae) used as the out-group, node labels indicate the posterior probability after 1 × 106 iterations.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Funding information
This work was supported by the Cotton Research and Development Corporation (CRDC) Australia.
References
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler F. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69:313–319.
- Coleman R, Hereward J, De Barro P, Frohlich D, Adamczyk J, Goolsby J. 2008. Molecular comparison of Creontiades plant bugs from south Texas and Australia. Southwestern Entomol. 33:111–117.
- Hereward J, Walter G. 2012. Molecular interrogation of the feeding behaviour of field captured individual insects for interpretation of multiple host plant use. PLoS One. 7:e44435.
- Hereward J, Walter G, Debarro P, Lowe A, Riginos C. 2013a. Gene flow in the green mirid, Creontiades dilutus (Hemiptera: Miridae), across arid and agricultural environments with different host plant species. Ecol Evol. 3:807–821.
- Hereward J, Debarro P, Walter G. 2013b. Resolving multiple host use of an emergent pest of cotton with microsatellite data and chloroplast markers (Creontiades dilutus Stål; Hemiptera, Miridae). Bull Entomol Res. 103:611–618.
- Hua J, Li M, Dong P, Cui Y, Xie Q, Bu W. 2008. Comparative and phylogenomic studies on the mitochondrial genomes of Pentatomomorpha (Insecta: Hemiptera: Heteroptera). BMC Genomics. 9:610.
- Huelsenbeck J, Ronquist F. 2001 . MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 17:754–755.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28:1647–1649.