
abstract
The complete mitogenome of the rocket frog Anomaloglossus baeobatrachus was sequenced using a shotgun approach on an Illumina HiSeq 2500 platform (Illumina Inc., San Diego, CA), providing the first mitogenome for this genus. The genome was 17,572 bp long and presents the typical organization found in other neobatrachian anurans. A phylogenetic analysis including A. baeobatrachus and all other available mitogenomes of Hyloidea provided relationships in accordance with previous phylogenetic studies.
Anomaloglossus baeobatrachus (Boistel & Massary, Citation1999) is a species of frog endemic to the eastern part of the Guiana Shield. It is currently known to occur in French Guiana, Suriname and the State of Amapá (Fouquet et al. Citation2012), and the State of Pará (Avila-Pires et al. Citation2010). The taxonomy of the genus Anomaloglossus is not well resolved, as several mitochondrial lineages currently associated with nominal species might in fact represent undescribed species (Fouquet et al. Citation2007, Citation2012; Kok et al. Citation2012). This is the case of A. baeobatrachus for which four distinct mitochondrial lineages have been identified (Fouquet et al. Citation2012). Molecular data can significantly contribute in resolving the systematics and species boundaries within this genus but available genomic data are still scarce. Here, we describe the complete mitochondrial genome of Anomaloglossus baeobatrachus.
A calling male of A. baeobatrachus was collected at Saint-Eugène, French Guiana (4°49'17.2''N; 53°04'03.4''W), the terra typica of the species (Boistel & Massary, personal communication). DNA was isolated from liver tissue using the Wizard Genomic extraction protocol (Promega Inc., Madison, WI). We then used 200 ng of DNA to create a DNA sequencing library at the Genopole of Toulouse (France). The library was hybridized and sequenced on a 1/24th of lane of an Illumina HiSeq 2500 flow cell (Illumina Inc., San Diego, CA). Over 24 million paired-end read of 150 bp were obtained. The mitochondrial genome was assembled using an iterative mapping strategy (Besnard et al. Citation2014). We obtained a circular sequence of 17,572 bp in length. The overall base composition was as follows: A (28.5%), C (27.4%), G (13.9%) and T (30.3). We annotated the mitogenome with the MITOS webserver (Bernt et al. Citation2013). We validated the coding regions using Geneious version 9.0.5 (Kearse et al. Citation2012). The annotated sequence was submitted to NCBI (accession no. KU958559).
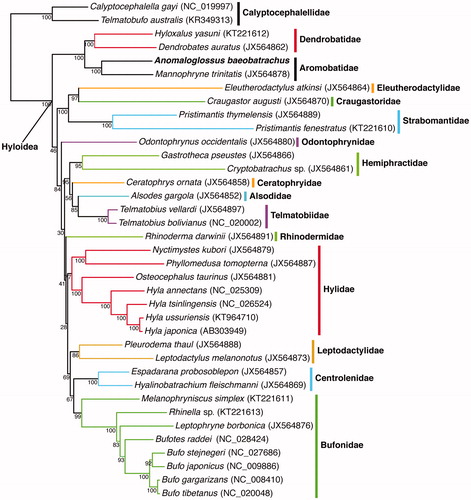
We then used MAFFT v.7 (Katoh & Standley Citation2013) to align the mitogenome of A. baeobatrachus with all available mitochondrial genomes of Hyloidea (Nobleobatrachia), a superfamily of Neobatrachia. The gene order was fully conserved in this clade, and we conducted a maximum-likelihood phylogenetic analysis on this alignment with RAxML v. 8.2.4. (Stamatakis Citation2014) excluding the control region. The resulting phylogenetic tree () shows that A. baeobatrachus and Mannophryne trinitatis, which belong to the family Aromobatidae, form a strongly supported clade. This clade is the sister group of Dendrobatidae, which is in accordance with previous studies (Grant et al. Citation2006). Given several species within this genus might face decline or might already have gone extinct (Courtois et al. Citation2015; Fouquet et al. Citation2015), resolving taxonomic uncertainties is crucial to assess conservation priorities. These data, which represent the first mitogenome for the genus and the second for Aromobatidae, will serve as a reference for further studies on the taxonomy and evolution of this group of amphibians.
Figure 1. Maximum-likelihood phylogeny of Hyloidea inferred with a GTR + G + I model from all available mitochondrial genomes in this clade. Calyptocephalellidae was used to root the tree. The new sequence is represented in bold. The Bootstrap values (based on 1000 iterations and 100 independent maximum-likelihood searches) are indicated for each internal node.
Funding information
This work has benefited from an “Investissement d'Avenir” grant managed by Agence Nationale de la Recherche (CEBA, ref.ANR-10-LABX-25-01), France.
Acknowledgements
The authors would like to thank Benoît Villette, Jean-François Szpigel and Sébastien Cally for their help in the field, and Guillaume Besnard for his advices on genome assembling and suggestions to improve the manuscript.
Disclosure statement
The authors report that they have no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Related Research Data
References
- Avila-Pires TCS, Hoogmoed MS, da Rocha WA. 2010. Notes on the vertebrates of northern Pará, Brazil: a forgotten part of the Guianan Region, I. Herpetofauna. Bol Mus Para Emílio Goeldi Cienc Nat, Bélem. 5:13–112.
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69:313–319.
- Besnard G, Jühling F, Chapuis É, Zedane L, Lhuillier É, Mateille T, Bellafiore S. 2014. Fast assembly of the mitochondrial genome of a plant parasitic nematode (Meloidogyne graminicola) using next generation sequencing. CR Biol. 337:295–301.
- Boistel R, Massary JC. 1999. Les amphibiens vénéneux de la famille des dendrobatidés. Le Courrier de la Nature. 176:34–39.
- Courtois EA, Gaucher P, Chave J, Schmeller DS. 2015. Widespread occurrence of Bd in French Guiana, South America. PLoS One. 10:e0125128.
- Fouquet A, Gilles A, Vences M, Marty C, Blanc M, Gemmell NJ. 2007. Underestimation of species richness in neotropical frogs revealed by mtDNA analyses. PLoS One. 2:e1109.
- Fouquet A, Marques Souza S, Sales Nunes PM, Kok PJR, Curcio FF, de Carvalho CM, Grant T, Rodrigues MT. 2015. Two new endangered species of Anomaloglossus (Anura: Aromobatidae) from Roraima State, northern Brazil. Zootaxa. 3926:191–210.
- Fouquet A, Noonan BP, Rodrigues MT, Pech N, Gilles A, Gemmell NJ. 2012. Multiple quaternary refugia in the Eastern Guiana Shield revealed by comparative phylogeography of 12 frog species. Syst Biol. 61:461–489.
- Grant T, Frost DR, Caldwell JP, Gagliardo R, Haddad CFB, Kok PJR, Means DB, Noonan BP, Schargel WE, Wheeler WC. 2006. Phylogenetic systematics of dart-poison frogs and their relatives (Amphibia: Athesphatanura: Dendrobatoidea). Bull Am Mus Nat Hist. 299:1–262.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30:772–780.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28:1647–1649.
- Kok PJR, MacCulloch RD, Means DB, Roelants K, Van Bocxlaer I, Bossuyt F. 2012. Low genetic diversity in tepui summit vertebrates. Curr Biol. 22:R589–R590.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30:1312–1313.