
Abstract
Pterosagitta draco is an important predator in many food webs, and so maintaining the diversity study of Pterosagitta draco in marine ecosystem is essential. In this paper, the complete mitochondrial genome sequence of Pterosagitta draco was determined. Its complete mtDNA sequence is 10,426 bp in length, which contains 11 protein-coding genes (PCGs), 6 transfer RNA genes and 2 ribosomal RNA genes. The composition of A, T, G, C in mtDNA is 27.63%, 23.61%, 23.53% and 25.23%, respectively. The percentage of A + T is 51.24%. The complete mitochondrial genome sequence would be useful for further phylogenetic analysis in Pterosagitta draco.
Chaetognaths comprise the relatively isolated marine phylum of approximately 209 species recorded in the world’s oceans (Souza et al. Citation2014). Chaetognaths are abundant planktonic organisms and important predators in many marine food webs (Jennings et al. Citation2010). Pterosagitta draco is an oceanic, epiplanktonic, cosmopolitan species of warm temperate regions (Shimotori et al. Citation1997). Several mitochondrial genomes of chaetognaths have been reported previously (Papillon et al. Citation2004; Helfenbein et al. Citation2004; Faure & Casanova Citation2006; Miyamoto et al. Citation2010; Li et al. Citation2015), but the mitochondrial genomes of Pterosagitta draco have not been reported. In this study, we applied advanced sequencing technologies to exploit complete mitochondrial genome sequences of Pterosagitta draco, which could supplement the mitogenome database for Chaetognatha. The primary specimens had been deposited in the South China Sea Institute of Oceanology, Chinese Academy of Sciences, Guangzhou, China and the accession number is SCSMBC040496.
A total of 29 individuals of Pterosagitta draco was collected from the northern South China Sea (21°50′N, 119°59′E) in a depth of 200 m to the surface, on 12 August 2015. The collected samples were smashed and preserved in ethanol, genomic DNA was extracted with marine animal gDNA kit (Biomiga, GD3311-02, San Diego, CA). The genomic DNA was extracted from the total sample and was sequenced by Illumina’s HiSeq2000 platform (Illumina, San Diego, CA) with 200 bp insert size and a pair-end 100 bp sequencing strategy (Tan et al. Citation2015). The raws reads were then filtered according to the protocols as Zhou et al. (Citation2013) described previously. The SOAPdenovo-Trans was applied to assemble the remaining high-quality reads as Xie et al. (Citation2014) reported previously. The contigs and scaffolds collected from the assembler were then annotated for protein-coding genes (PCGs) by homolog prediction (Zhou et al. Citation2013), tRNAs by tRNAscan-SE by tRNAscan-SE (Schattner et al. Citation2005), rRNAs by Geneious 6.1.2 (Kearse et al. Citation2012). Four previously published mitogenomes of Chaetognatha (Sagitta nagae, Sagitta decipiens, Sagitta ferox and Sagitta enflata) on NCBI were further used to determine the boundaries of genes (Miyamoto et al. Citation2010; Li et al. Citation2015).
The complete mtDNA sequence of Pterosagitta draco is 10,426 bp in length; it contains 11 PCGs, 6 transfer RNA genes and 2 ribosomal RNA genes. The composition of A, T, G, C in mtDNA is 27.63%, 23.61%, 23.53% and 25.23%, respectively. The annotated mitogenome of Pterosagitta draco has been submitted to GenBank (Accession number is KU507531).
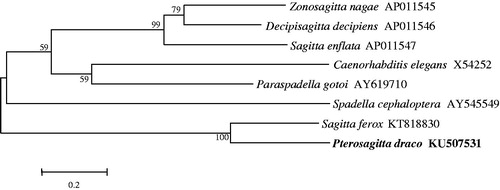
The complete mtDNA sequence of Pterosagitta draco was further applied to generate the phylogram with complete mitogenomes of closely related Chaetognatha. ClustalW was used to perform the multiple sequence alignments (Zhou et al. Citation2016). The dendrogram was calculated by the Neighbor Joining method and MEGA 5 software (Arizona State University, Tempe, AZ, USA) (Saitou & Nei Citation1987; Tamura et al. Citation2011) (). The complete mitogenome of Pterosagitta draco will provide important information in phylogenetic analysis at species and family level.
Figure 1. The phylogram generation according to complete mitogenomes. The dendrogram was established by the Neighbor Joining method and MEGA 5 software. Numbers at each node indicate bootstrap support.
Acknowledgements
We are grateful to the captain, crew and scientists of R/V Shiyan 3, South China Sea Institute of Oceanology, Chinese Academy of Sciences, for their helpful assistance in collecting the zooplankton samples. Special thanks are due to Dr. Kaizhi Li of South China Sea Institute of Oceanology for her help in coordinating the sampling and relevant project.
Disclosure statement
The authors report no conflict of interest and are responsible for the content and writing of the paper.
Funding information
This work was supported by Strategic Priority Research Program of the Chinese Academy of Sciences (XDA11020305).
Related Research Data
References
- Faure E, Casanova JP. 2006. Comparison of Chaetognath mitochondrial genomes and phylogenetical implications. Mitochondrion. 6:258–262.
- Helfenbein KG, Fourcade HM, Vanjani RG, Boore JL. 2004. The mitochondrial genome of Paraspadella gotoi is highly reduced and reveals that chaetognaths are a sister group to protostomes. Proc Natl Acad Sci U S A. 101:10639–10643.
- Jennings RM, Bucklin A, Pierrot-Bults A. 2010. Barcoding of arrow worms (Phylum Chaetognatha) from three oceans: genetic diversity and evolution within an enigmatic phylum. PLoS One. 5:e9949.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28:1647–1649.
- Li P, Yang M, Ni S, Zhou L, Wang Z, Wei S, Qin Q. (2015). Complete mitochondrial genome sequence of the pelagic chaetognath, sagitta ferox. Mitochondrial DNA. 18:1–2.
- Miyamoto H, Machida RJ, Nishida S. 2010. Complete mitochondrial genome sequences of the three pelagic chaetognaths Sagitta nagae, Sagitta decipiens and Sagitta enflata. Comp Biochem Physiol Part D Genomics Proteomics. 5:65–72.
- Papillon D, Perez Y, Caubit X, Le Parco Y. 2004. Identification of Chaetognaths as protostomes is supported by the analysis of their mitochondrial genome. Mol Biol Evol. 21:2122–2129.
- Saitou N, Nei M. 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 4:406–425.
- Schattner P, Brooks AN, Lowe TM. 2005. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33:W686–W689.
- Shimotori T, Goto T, Terazaki M. 1997. Egg colony and early development of Pterosagitta draco (Chaetognatha) collected from Kuroshio front. Plankton Biol Ecol. 44:71–80.
- Souza CS, Luz JA, Mafalda PO. 2014 . Relationship between spatial distribution of chaetognaths and hydrographic conditions around seamounts and islands of the tropical southwestern Atlantic. An Acad Bras Cienc. 86:1151–1165.
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 28:2731–2739.
- Tan M, Zhang R, Hardman C, Zhou X. (2015). Mitochondrial genome of Hylaeus dilatatus (Hymenoptera: Colletidae). Mitochondrial DNA. 28:1–2.
- Xie Y, Wu G, Tang J, Luo R, Patterson J, Liu S, Huang W, He G, Gu S, Li S, et al. 2014. SOAPdenovo-Trans: de novo transcriptome assembly with short RNA-Seq reads. Bioinformatics. 30:1660–1666.
- Zhou C, Tan M, Du S, Zhang R, Machida R, Zhou X. (2016). The mitochondrial genome of the winter stonefly Apteroperla tikumana (Plecoptera, Capniidae). Mitochondrial DNA A DNA MappSeq Anal. 27:3030–3032.
- Zhou X, Li Y, Liu S, Yang Q, Su X, Zhou L, Tang M, Fu R, Li J, Huang Q. 2013. Ultra-deep sequencing enables high-fidelity recovery of biodiversity for bulk arthropod samples without PCR amplification. Gigascience. 2:4.