
Abstract
The mitogenome of Hyalella lucifugax from Lake Titicaca, obtained using Illumina NGS technology, is described. The mitogenome attains 14,994 bp (although the control region could not be completed) and comprises the standard set of 2 rRNAs, 13 protein-coding genes, and 22 tRNA genes, plus two non-coding regions. A phylogenetic analysis based on the protein-coding mitochondrial genes from representatives from all amphipod genera with available sequences in GenBank recovers the monophyly of H. lucifugax with the superfamily Talitroidea.
Hyalella Smith, 1874, is the most common amphipod genus in inland waters of both North and South America, and constitutes a model organism in ecotoxicology and gene expression studies (Major et al. Citation2013; Wu et al. Citation2014; Hasenbein et al. Citation2015). The genus – endemic to the Holarctic and Neotropical regions – currently comprises 70 species (Horton & Lowry Citation2013), of which 18 occur in Lake Titicaca, most of them being endemic to the Lake (González & Coleman Citation2002). Here, we present the first mitogenome sequence for the genus Hyalella. The H. lucifugax (Faxon, 1876) sample was collected in 2007 at 23 m depth in Bahía de Copacabana, on the Peruvian sector of the lake (16°12′10.80″S, 69°23′27.60″W). Voucher specimens have been deposited at the DNA and tissue collection of the Biodiversity, Systematics, and Evolution group (Bio6Evo) of the University of the Balearic Islands with accession number 1618.
A shotgun genomic library was sequenced in parallel with other samples on the Illumina HiSeq2500 platform version 3 (Illumina Inc.) with 160 bp paired-end reads using standard protocols. A nearly complete mitogenome of 14,994 bp (6362 high-quality reads, average coverage 68×) was obtained. Genes were annotated using the MITOS webserver (Bernt et al. Citation2013) and sequenced with the annotated features submitted to ENA (accession number LT594767).
The mitogenome shows an A + T bias (A 33.73%; C 14.09%; G 15.47%; and T 36.71%) and the gene content comprising 13 protein-coding genes (PCGs), 22 tRNAs, and 2 ribosomal genes. An AT-rich (85.65%) non-coding region of at least 223 bp (but that might be longer since the region could not be completed) is located between rrnS and trnC genes. This region can potentially form a stem-loop secondary structure that might correspond to the origin of replication. Another downstream non-coding region of 663 bp is also AT-rich (85.07%) and includes numerous short palindromes. PCGs order is identical to the putative ancestral Pancrustacean mitogenome but some tRNA genes seem to have been rearranged. Most genes are encoded at the + strand (with the exception of nad1, nad4, nad4l, nad5, rrnS, and rrnL and seven tRNAs). Start codons correspond to the canonical ATA or ATG except for cox1 and nad4L (ATT), atp8 (ATC), and nad5 (TTG). Likewise, most stop-codons are the canonical TAA or TAG except for nad2, nad5, and nad4 genes that instead display a single T.
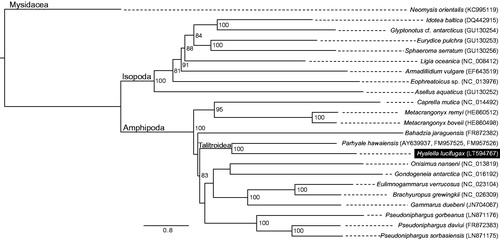
Representative species from all amphipod genera with mitogenome sequences available in GenBank plus isopods and a mysid as outgroup were used to assess the phylogenetic placement of H. lucifugax, which was recovered as sister to Parhyale hawaiensis (Dana, 1853) (superfamily Talitroidea; ).
Figure 1. Maximum-likelihood phylogeny obtained using IQTREE multicore v. 1.3.12 (Nguyen et al. Citation2015) based on the nucleotide sequences of protein-coding genes of 14 amphipod species plus nine outgroups showing the systematic placement of H. lucifugax. The nucleotide sequences of the 13 PCGs were aligned in TranslatorX (Abascal et al. Citation2010) with poorly aligned regions removed in Gbloks v. 0.91b (Talavera & Castresana Citation2007). The best partitioning scheme and evolutionary model for each partition was assessed in PartitionFinder v 1.1.1 (Lanfear et al. Citation2012). Node numbers represent bootstrap support values evaluated with 1000 bootstrap replicates. GenBank accession numbers follow species names.
Disclosure statement
The authors report no conflicts of interest and are responsible for the content and writing of the article.
Acknowledgements
We thank Oliver Kroll, Christian Albrecht and Tom Wilke (Department of Animal Ecology and Systematics, Justus Liebig University Giessen, Germany) for kindly donating tissue samples used in this study.
References
- Abascal F, Zardoya R, Telford MJ. 2010. TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 38:W7–13.
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69:313–319.
- González E, Coleman CO. 2002. (Crustacea, Amphipoda, Hyalellidae) and the description of a related new species from Lake Titicaca. Org Divers Evol. 2:271–273.
- Hasenbein S, Connon RE, Lawler SP, Geist J. 2015. A comparison of the sublethal and lethal toxicity of four pesticides in Hyalella azteca and Chironomus dilutus. Environ Sci Pollut Res Int. 22:11327–11339.
- Horton T, Lowry J. 2013. Hyalella S I Smith, 1874 In: Horton T, Lowry J, De Broyer C, Bellan-Santini D, Coleman CO, Daneliya M, Dauvin J-C, Fišer C, Gasca R, Grabowski M, et al., editors. 2016. World Amphipoda Database. Available from: http://www.marinespecies.org/aphia.php?p=taxdetails&id =158104.
- Lanfear R, Calcott B, Ho SYW, Guindon S. 2012. Partitionfinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol. 29:1695–1701.
- Major K, Soucek DJ, Giordano R, Wetzel MJ, Soto-Adames F. 2013. The common ecotoxicology laboratory strain of Hyalella azteca is genetically distinct from most wild strains sampled in eastern North America. Environ Toxicol Chem. 32:2637–2647.
- Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32:268–274.
- Talavera G, Castresana J. 2007. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 56:564–577.
- Wu YH, Wu TM, Hong CY, Wang YS, Yen JH. 2014. Screening differentially expressed genes in an amphipod (Hyalella azteca) exposed to fungicide vinclozolin by suppression subtractive hybridization. J Environ Sci Heal Part B. 49:856–863.