
Abstract
The dusky-footed woodrat (Neotoma fuscipes) is an endemic North American rodent belonging to the family Cricetidae. We present here the first complete mitogenome within the Neotoma genus. The mitogenome is 16,199 bp in length, and has a structure and gene organization similar to other rodent species (e.g. Mus musculus, Peromyscus maniculatus and Microtus fortis calamorum). The overall base composition is A (35.2%), C (25.5%), G (12.3%), T (27.0%), with a GC content of 37.8%, similar to other rodents. This mitogenome serves as a foundation for future phylogenetic, phylogeographic and population genetic studies.
The 22 species of woodrats within the genus Neotoma are widespread throughout North America (Nowak Citation1999). They are important for paleoecological studies, serving as paleothermometers (Smith & Betancourt Citation1998) and as collecting agents for Quaternary fossils (Betancourt et al. Citation1990). A complete understanding of the genetic makeup of the species within Neotoma would thus facilitate future studies using both modern and ancient DNA. Here, we present the first mitogenome description within the genus Neotoma, which will help further resolve the evolutionary history of this species, and its relationship with other cricetid rodents.
We sequenced two dusky-footed woodrat (Neotoma fuscipes) individuals from Samwell Cave, CA (MVZ:Mamm:225109; longitude/latitude (−122.239893, 40.9176)) and the Snow Mountain Wilderness, CA (MVZ:Mamm:196404; longitude/latitude (−122.66451, 39.3450164)). DNA was extracted from muscle tissue via the Qiagen DNeasy kit (Qiagen, Valencia, CA). The complete mitochondrial genome was created through genomic library preparation using the NEBNext Ultra DNA kit (New England Biolabs, Ipswich, MA), and subsequently sequenced on a single HiSeq2000 lane (Illumina, San Diego, CA). We aligned our genomic short reads against Mus musculus (GenBank accession number NC_005089.1) and Peromyscus maniculatus (Hawkins et al. Citation2016) mitogenomes with BWA (v0.7.12, Li & Durbin Citation2009) and Samtools (v1.3, Li et al. Citation2009). These reads were then aligned to Microtus fortis calamorum (Genbank accession: JF261175.1) in Geneious (v8.1.8, Biomatters, Auckland, New Zealand). The mitogenome was annotated by the programs DOGMA (Wyman et al. Citation2004) and MITOS (Bernt et al. Citation2013), and tRNA inconsistencies were resolved with tRNAscan-SE (Lowe & Eddy Citation1997). We used IQtree v1.4.3 (Trifinopoulos et al. Citation2016) to determine the best model of evolution amongst N. fuscipes and 17 other Cricetid mitogenomes, and reconstructed their phylogenetic relationships using Sciurus vulgaris as the outgroup.
The Neotoma fuscipes mitogenome (GenBank accession number KU745736) has a total length of 16,199 bp (average coverage of 3129×). The overall base composition is A (35.2%), C (25.5%), G (12.3%), T (27.0%), with a GC content of 37.8%, similar to other rodents (Partridge et al. Citation2007). The mitogenome arrangement is also similar to other mammals with 13 protein-coding genes, 2 rRNA genes and 22 transfer RNAs. Gene order and organization is similar to Mus musculus (NC_005089.1) and Microtus fortis calamorum (Genbank accession number JF261175.1). Annotation with DOGMA (Wyman et al. Citation2004) and MITOS (Bernt et al. Citation2013) provided fairly consistent results. tRNAscan-SE did not identify two tRNAs that were found with both MITOS and DOGMA: tRNA-VAL (1027-1097) and tRNA-SER (11606-11663). These tRNAs were added to the final annotation.
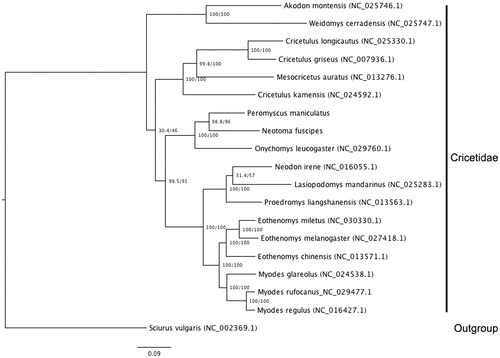
Model testing in IQtree revealed TIM2 + I + G4 to be the best-supported model of evolution. The resulting phylogeny places Onychomys, Neotoma and Peromyscus in a single clade, with Neotoma sister to Peromyscus (). This placement contrasts with previous phylogenies based on one or multiple mitochondrial and/or nuclear genes (e.g. Bradley et al. Citation2004; Reeder et al. Citation2006), underscoring the uncertain placement of these taxa within Neotominae, though we note that full mitogenomes are available for only a few species. The mitogenome of Neotoma fuscipes will thus be useful for future phylogenetic, phylogeographic and population genetic studies.
Figure 1. Maximum likelihood (ML) tree based on complete mitogenomes from the family Cricetidae, under the model TIM2 + I + G4. Bootstrap support at nodes depicts bootstraps in front of the diagonal (/) and ultra-fast bootstraps, after the diagonal. The number in parentheses following each specimen is the NCBI accession number, except in the case of Peromyscus maniculatus (from Hawkins et al. Citation2016) and Neotoma fuscipes (this study).
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper. The complete mitochondrial DNA sequence produced in this paper has been deposited in NCBI GenBank under accession number KU745736.
Acknowledgments
We thank the UC Berkeley Museum of Vertebrate Zoology, Marjorie Matocq, Jim Patton, and Chris Conroy for their extensive field collections and specimen identifications of Neotoma fuscipes.
This work used the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley, supported by NIH S10 Instrumentation Grants S10RR029668 and S10RR027303.
References
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69:313–319.
- Betancourt JL, Van Devender TR, Martin PS. 1990. Packrat middens. Tucson (AZ): University of Arizona Press.
- Bradley RD, Edwards CW, Carroll DS, Kilpatrick CW. 2004. Phylogenetic relationships of neotomine-peromyscine rodents: based on DNA Sequences from the mitochondrial cytochrome-b gene. J Mammal. 85:389–395.
- Hawkins MTR, Hofman CA, Callicrate T, McDonough MM, Tsuchiya MTN, Gutierrez EE, Helgen KM, Maldonado JE. 2016. In solution hybridization for mammalian mitogenome enrichment: pros, cons, and challenges associated with multiplexing degraded DNA. Mol Ecol Resour. 16:1173–1188.
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-wheeler transform. Bioinformatics. 25:1754–1760.
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The sequence alignment/map format and SAMtools. Bioinformatics. 25:2078–2079.
- Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25:955–964.
- Nowak R. 1999. Walker's mammals of the world, vol. 2, 6th ed. Baltimore: The Johns Hopkins University Press.
- Partridge MA, Davidson MM, Hei TK. 2007. The complete nucleotide sequence of Chinese hamster (Cricetulus griseus) mitochondrial DNA. DNA Sequence. 18:341–346.
- Reeder SA, Carroll DS, Edwards CW, Kilpatrick CW, Bradley RD. 2006. Neotomine–peromyscine rodent systematics based on combined analyses of nuclear and mitochondrial DNA sequences. Mol Phylogenet Evol. 40:251–5258.
- Smith F, Betancourt J. 1998. Response of bushy-tailed woodrats (Neotoma cinerea) to late Quaternary climatic change in the Colorado Plateau. Quatern Res. 50:1–11.
- Trifinopoulos J, Nguyen LT, von Haeseler A, Minh BQ. 2016. W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 44: W232–W235.
- Wyman SK, Jansen RK, Boore JL. 2004. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 20:3252–3255.