
Abstract
We describe six novel ungulate-specific conserved primers for sequencing the complete mitochondrial cytochrome c oxidase I (COI) gene of selected threatened species from degraded samples for effective conservation planning. These primers amplified 301–1599 bp DNA fragments in various combinations. The method described may assist in the sequencing of either complete gene from moderate to good quality DNA or shorter fragment from degraded DNA.
Introduction
Terrestrial mammals are threatened due to extinction risk, and around 50% of them are showing a declining trend (Channell & Lomolino Citation2000; Ceballos et al. Citation2005). Among these, the South Asian mammals are the most endangered group (Schipper et al. Citation2008), specially the ungulates, which are declining due to environmental changes, impacts of anthropogenic pressure on wildlife habitats and poaching (Estes et al. Citation2006). Conservation success largely depends upon identifying vulnerable species and understanding the environmental factors that support their persistence in human-dominated landscapes. In recent years, molecular taxonomy has helped in resolving the phylogeny of cervids (Pitra et al. Citation2004; Gilbert et al. Citation2006) resulting in clarity on species distribution and relatedness for effective conservation planning. However, studies indicate further revision in the molecular phylogeny (Groves & Grubb Citation2011; Timmins et al. Citation2015). Successful conservation efforts depend upon the identification of evolutionary significant units (ESU) of vulnerable species. The mitochondrial DNA (mtDNA) cytochrome c oxidase I (COI) gene has been widely used for biological identification and phylogenetic studies (Weibel & Moore Citation2002; Hebert et al. Citation2003). Further enriching of its database with the DNA sequences of COI gene would be an asset for the identification of ESU of vulnerable species exhibiting declining trend and reduced genetic variations. It requires large amounts of sequence data from different populations.
With increasingly stringent wildlife laws (e.g. Indian Wildlife (Protection) Act, 1972), obtaining permission for collection of invasive biological samples is difficult. Therefore, researchers often utilize decomposed and non-invasively collected biological samples for addressing the genetic issue. Amplification and sequencing of a complete gene from the DNA extracted from such samples is a challenging task. With our experience of working on population genetics and molecular phylogeny of swamp deer (Rucervus duvaucelii), sambar (Rusa unicolor), hog deer (Axis porcinus), wild pig (Sus scrofa) and other ungulates (Gupta et al. Citation2013a; Angom et al. Citation2015), we describe novel primers for sequencing the complete mtDNA COI gene by amplification of shorter fragment from the DNA extracted from non-invasive and degraded samples.
Complete COI sequences with the flanking region of various ungulate species were obtained from GenBank and aligned using ClustalW (Thompson et al. Citation1994). After alignment of the sequences, the homologous regions were selected for designing three pairs of conserved primer ().
Table 1. Six primers designed for amplification of either complete or partial fragment of mtDNA COI gene.
We used biological samples collected from the field for validation of primers. DNA was extracted (Gupta et al. Citation2013b) and subjected to PCR amplification using different primer combinations (). A range of annealing temperature was tested for multiplex amplification, and optimum amplification was obtained at 56 °C. PCR amplification was carried out in a 20 μl reaction volume containing 1 μl of the extracted DNA, 4 pmol of each primer and 1× Multiplex PCR Kit (Qiagen, Hilden, Germany). We performed PCR in the following steps: initial denaturation at 95 °C for 15 min, followed by 35 cycles of denaturation at 95 °C for 45 seconds, annealing at 56 °C for 1 min, and extension at 72 °C for 1.5 min. Final extension was at 72 °C for 10 min. We used 2.2% agarose gel containing ethidium bromide to visualize PCR products under a UV transilluminator (). These were sequenced from both the strands using ABI 3130 Genetic Analyser. Sequences were aligned using SeqScape v2.5 (Applied Biosystems, Foster City, CA). A neighbour-joining phylogenetic tree was constructed using the Kimura 2-parameter method (Kimura Citation1980) with MEGA 7 (Kumar et al. Citation2016).
Figure 1. Image of the agarose gel showing amplification result of multiplex PCR reaction with the DNA extracted from partially decomposed tissue of a verity of ungulate species as labelled at each lane. (A) PCR amplification with F1, R1, R2 and R3 primers; (B) amplification with F1, F2, R2 and R3 primers; (C) amplification with F1, F2, R2 and R3 and (D) amplification with F1, F2, F3 and R3. Numeric are the base pair size of respective amplicons.
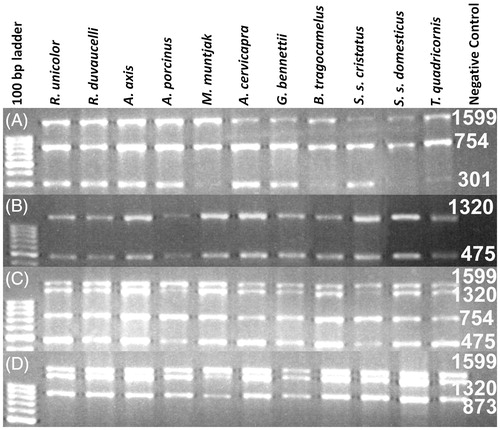
Table 2. All possible combinations of the primers during PCR amplification with tested samples and expected amplicon lengths.
Results and conclusions
Expected PCR amplicons were obtained with different primer combinations ( and ). Less than 1 KB fragments (301, 475, 754, and 837 bp) were amplified with the DNA extracted from all the biological samples excluding faecal samples (). The maximum amplicon size from the faecal DNA was 475 bp. PCR products of 1320 and 1599 bp were amplified from the DNA extracted from fresh and less decomposed tissue samples (). A complete gene consensus sequence was obtained for various tested ungulate species and deposited in the NCBI GenBank (KT372090-2100), as shown in . The neighbour-joining (NJ) tree exhibited the expected phylogenetic relationship among the tested species ().
Figure 2. Phylogenetic tree based on neighbour-joining method using MEGA 7. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) are shown next to the branches.
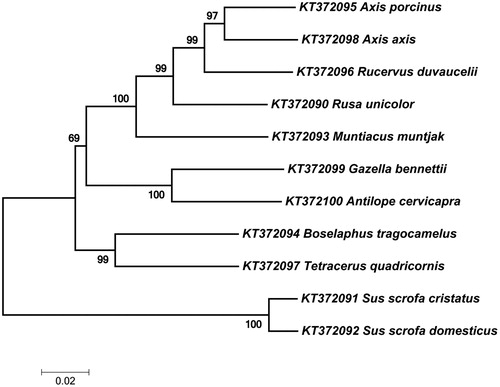
Table 3. List of ungulate species used for validation of primers.
Availability of virtue of DNA sequences from a different population of species facilitates the improved phylogenetics. For the collection of invasive samples, immobilization of wild animal using tranquilization drugs involves risk on their life. However, tranquilization-free collection of samples from the endangered wild animal is an efficient way of increasing the sample size. Therefore, amplification of smaller fragments to generate a complete gene sequence is a convenient way of enhancing databases for those species when invasive samples are difficult to obtain. The availability of complete sequences for multiple genes from a variety of species can improve the reliability of sequence-based species identification and phylogeny. The primer combinations described here will be beneficial for amplification and sequencing of complete COI gene of ungulate species using decomposed biological samples. The COI sequence along with widely deployed cytochrome b gene can be used to generate the unambiguous phylogenic tree.
Acknowledgments
This study was funded by the WII and Scientific & Engineering Research Board, Department of Science and Technology, Govt. of India (Grant number SR/SO/AS-77/2013). We acknowledge the support of the Director, Dean and Nodal Officer (Wildlife Forensic and Conservation Genetics Cell) of the WII.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Additional information
Funding
Related Research Data
References
- Angom S, Gupta SK, Kumar A, Hussain SA. 2015. Identification of globally threatened cervids from problematic samples using cytochrome b and control region genes. Conserv Genets Resour. 7:647–650.
- Ceballos G, Ehrlich PR, Soberon J, Salazar I, Fay JP. 2005. Global mammal conservation: what must we manage? Science. 309:603–607.
- Channell R, Lomolino M. 2000. Dynamic biogeography and conservation of endangered species. Nature. 403:84–86.
- Estes RD, Atwood JL, Estes AB. 2006. Downward trends in Ngorongoro Crater ungulate populations 1986–2005: conservation concerns and the need for ecological research. Biol Conser. 131:106–120.
- Gilbert C, Ropiquet A, Hassanin A. 2006. Mitochondrial and nuclear phylogenies of Cervidae (Mammalia, Ruminantia): systematics, morphology, and biogeography. Mol Phylogenet Evol. 40:101–117.
- Groves C, Grubb P. 2011. Ungulate taxonomy. Baltimore, USA: The Johns Hopkins University Press.
- Gupta SK, Kumar A, Hussain SA. 2013a. Extraction of PCR-amplifiable DNA from a variety of biological samples with uniform success rate. Conserv Genets Resour. 5:215–217.
- Gupta SK, Kumar A, Hussain SA, Vipin Singh L. 2013b. Cytochrome b based genetic differentiation of Indian wild pig (Sus scrofa cristatus) and domestic pig (Sus scrofa domestica) and its use in wildlife forensics. Sci Just. 53:220–222.
- Hebert PDN, Cywinska A, Ball SL, deWaard JR. 2003. Biological identifications through DNA barcodes. Proc Biol Sci. 270:313–321.
- Kimura M. 1980. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 16:111–120.
- Kumar S, Stecher G, Tamura K. 2016. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 33:1870–1874.
- Pitra C, Fickela J, Meijaard E, Groves PC. 2004. Evolution and phylogeny of old world deer. Mol Phylogenet Evol. 33:880–895.
- Schipper J, Chanson JS, Chiozza F, et al. 2008. The status of the world’s land and marine mammals: diversity, threat, and knowledge. Science. 322:225–230.
- Thompson JD, Higgins DG, Gibson TJ. 1994. Clustal W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673–4680.
- Timmins R, Kawanishi K, Giman B, Lynam A, Chan B, Steinmetz R, Sagar BH, Kumar SN. 2015. Rusa unicolor. The IUCN Red List of Threatened Species. Version 2015.2 [Internet]. [cited 2015 Sep 1]. Available from: http://www.iucnredlist.org/details/41790/0
- Weibel AC, Moore WS. 2002. Molecular phylogeny of a cosmopolitan group of woodpeckers (Genus Picoides) based on COI and cyt b mitochondrial gene sequences. Mol Phyl Evol. 22:65–75.