
Abstract
Here, we present complete mitochondrial genome of the Eastern Slow Worm, Anguis colchica (Nordmann, 1840). Mitogenome complete sequence is 17,097 bp long and consists of 13 protein-coding genes, 22 tRNA genes, two rRNA genes and one control region. Anguis colchica mitochondrial genome has the same gene order as other mitogenomes of Anguis spp. Their analyzed genome has base composition as: A (30.4%), T (24.6%), C (30.4%), G (14.6%), with an A + T bias (55%). Length of the all 22 tRNA genes varies from 65 to 73 bp with an average of 69 bp. Presented mitogenome will provide new data for phylogenetic analysis within the genus Anguis.
Genus Anguis is a small group of lizards from Anguidae family called slow worms. Recently, these legless lizards were under a very intensive study, which resulted with its taxonomic revisions (Gvoždík et al. Citation2010; Gvoždík et al. Citation2013). Based on the analyses of both mitochondrial and nuclear DNA fragments, Gvoždík et al. (Citation2010) divided A. fragilis sensu lato into A. fragilis Linnaeus, 1758, A. colchica (Nordmann, 1840) and A. graeca Bedriaga, 1881 (Gvoždík et al. Citation2010). Furthermore, the same genetic tools proved the presence of fifth slow worm species, A. veronensis Pollini, 1818, endemic for Apennine Peninsula (Gvoždík et al. Citation2013). Further analyses confirmed earlier studies and showed distinctness of the new species and their deep intraspecific variability (Mezzasalma et al. Citation2013; Jablonski et al. Citation2016).
Although A. colchica originated by split from A. fragilis sensu lato (former subspecies; see Gvoždík et al. Citation2010) it belongs to relatively slightly known species. Although genetic data clearly separated Anguis spp. into five taxa, morphological differences did not confirm these findings (Gvoždík et al. Citation2010; Gvoždík et al. Citation2013). According to Gvoždík et al. (Citation2013) and Jablonski et al. (2016) the species occurs mainly in eastern parts of Europe, northern Anatolia, western Russia, Transcaucasian region and northern Iran. Thus, to provide more comprehensive data for phylogenetic analyses, we sequenced complete mitochondrial DNA (MtDNA) genome of A. colchica representative.
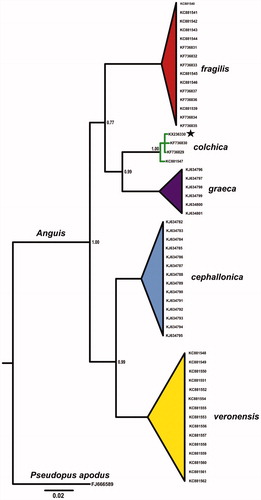
Total genomic DNA was isolated from a road-killed individual (adult male collected in Zatwarnica village, the Bieszczady Mts.; SE Poland; 49°13’47.5" N 22°33’20.1" E) with Sherlock AX (A&A Biotechnology, Gdynia, Poland) according to producers instructions. Analysed individual is deposited in the Faculty of Biological Sciences collection under voucher number AC 148, at the University of Zielona Góra. MtDNA complete genome was amplified with three overlapping fragments using PCR and then sequenced. Sequencing was carried out by Wyzer Biosciences (Cambridge, MA) using primer walking method. Genome assembling was done using MITOS WebServer (Bernt et al. Citation2013) and checked manually. The sample taxonomical status was confirmed with phylogenetic tree created with Bayesian inference using MrBayes 3.2.5 () (Ronquist et al. Citation2012).
Figure 1. Bayesian phylogenetic tree of Anguis spp. representatives, created with 765 bp of ND2 gene alignment. The tree was created using GTR + I +G model of substitution, as suggested by jModelTest 2.1.10 (Guindon & Gascuel Citation2003; Darriba et al. Citation2012). Tree was generated with 10,000,000 MCMC generations and 25% of burn-in. The individual used for complete mitochondrial genome is marked by star. A homologous sequence of Pseudopus apodus (Pallas, 1775) was used as outgroup. GenBank accession numbers and Bayesian posterior probabilities of nodes are shown on the tree.
The sequence of A. colchica complete mitochondrial genome had 17,097 bp (KX236330) and revealed the same gene order as other representatives of Anguis spp. Analysed genome was shorter than mtDNA of A. cephallonica (17,208 bp) and A. fragilis (17,479 bp). When compared to A. fragilis mitogenome, A. colchica had the same start and stop codons for all the protein-coding genes (PCGs), while comparison with A. cephallonica showed differences in one start codon (ND3 gene: GTG in A. colchica vs. ATG in A. cephallonica) and five stop codons (ND1 (TAA), ND2 (TAG), COX1 (TAG), ATP6 (TAX), ND5 (TXX) for A. colchica vs. ND1 (TAG), ND2 (TAA), COX1 (TAA), ATP6 (TAA), ND5 (TAA) for A. cephallonica). In A. colchica mitogenome four PCGs have used truncated stop codons (COX2, COX3, ND3, ND4). The analysed species H-strand overall base composition is as follows: A (30.4%), T (24.6%), C (30.4%), G (14.6%), with an A + T bias (55%).
Acknowledgements
DJ was supported by the grants VEGA 1/0073/14, UK/20/2014 and UK/37/2015. This work was supported by the Slovak Research and Development Agency under the contract no. APVV-0147-15 and by Wroclaw Centre of Biotechnology, program ‘The Leading National Research Centre (KNOW) of Wroclaw University of Environmental and Life Sciences for years 2014–2018’.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Additional information
Funding
References
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69:313–319.
- Darriba D, Taboada GL, Doallo R, Posada D. 2012. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 9:772.
- Guindon S, Gascuel O. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 52:696–704.
- Gvoždík V, Benkovský N, Crottini A, Bellati A, Moravec J, Romano A, Sacchi R, Jandzik D. 2013. An ancient lineage of slow worms, genus Anguis (Squamata: Anguidae), survived in the Italian Peninsula. Mol Phylogenet Evol. 69:1077–1092.
- Gvoždík V, Jandzik D, Lymberakis P, Jablonski D, Moravec J. 2010. Slow worm, Anguis fragilis (Reptilia: Anguidae) as a species complex: Genetic structure reveals deep divergences. Mol Phylogenet Evol. 55:460–472.
- Jablonski D, Jandzik D, Mikulíček P, Džukić G, Ljubisavljević K, Tzankov N, Jelić D, Thanou E, Moravec J, Gvoždík V. 2016. Contrasting evolutionary histories of the legless lizards slow worms (Anguis) shaped by the topography of the Balkan Peninsula. BMC Evol Biol. 16:99.
- Mezzasalma M, Guarino FM, Aprea G, Petraccioli A, Crottini A, Odierna G. 2013. Karyological evidence for diversification of Italian slow worm populations (Squamata, Anguidae). Comp Cytogenet. 7:217–227.
- Ronquist F, Teslenko M, Van Der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61:539–542.