
Abstract
In this paper, we present complete mitochondrial genome of the Italian legless lizard species Anguis veronensis Pollini, 1818. The complete mtDNA consisted of 13 protein-coding genes, 22 tRNAs, and two rRNA genes which in total formed a DNA strand of 17,322 bp. Anguis veronensis mitogenome had the same gene order as two other compared Anguis spp., i.e. A. cephallonica and A. fragilis. The base composition of A. veronensis mitochondrial genome was A – 30.8%, T – 24.9%, C – 29.9%, G – 14.4%, with an A + T bias (55.7%). The newly described genome provides valuable data for future comparative mitogenomic analysis within Anguis genus.
The genus Anguis (slow worms) comprising a group of Palearctic legless lizards, has recently been the subject of numerous genetic studies (Gvoždík et al. Citation2010; Gvoždík et al. Citation2013; Mezzasalma et al. Citation2013). Four out of five currently described species occur in the Balkans and only two of them (A. fragilis, A. colchica) have a broad distributions (Jablonski et al. Citation2016). South European endemic Anguis veronensis Pollini, 1818, is mainly distributed, although it is not entirely restricted, to the Italian Peninsula. The species has been recently resurrected based on molecular (Gvoždík et al. Citation2013) and karyological (Mezzasalma et al. Citation2013) markers, despite its weak morphological differentiation from other slow-worm species (Gvoždík et al. Citation2013). The phylogenetic relationships within the genus Anguis are still largely unresolved (Gvoždík et al. Citation2013). Therefore, we sequenced the complete mitochondrial genome (mtDNA) of this ancient Anguis lineage to provide a valuable new tool to resolve phylogenetic relationships (Douglas et al. Citation2006; Douglas & Gower Citation2010; Nabholz et al. Citation2013). Data obtained in this study, along with already sequenced mitochondrial genomes of Anguis species (Albert et al. Citation2009; Strzała et al. Citation2016), will provide opportunity of comprehensive mitogenomic study of these legless lizards.
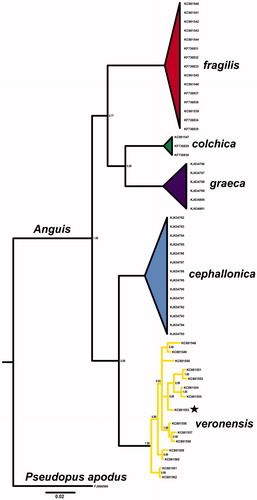
A tissue sample was collected from a road-killed individual in Bianzano (province of Bergamo; 45.774119°N, 9.921307°E) and is deposited in Zoological Collection of Angelica Crottini (ACZC3305) housed in CIBIO/InBIO, Research Centre in Biodiversity and Genetic Resources. ND2 sequence of this sample was already available and confirmed its taxon identity, which was also confirmed with Bayesian phylogenetic tree created with MrBayes 3.2.5 (Ronquist et al. Citation2012) (). (, KC881553). Total genomic DNA was isolated with Sherlock AX (A&A Biotechnology, Gdynia, Poland) according to the product manual. Mitochondrial genome was amplified with three overlapping fragments and sequenced using primer walking method by Wyzer Biosciences Inc. (Cambridge, MA). Mitochondrial genome was assembled with MITOS WebServer (Bernt et al. Citation2013) and checked manually.
Figure 1. Bayesian phylogenetic tree of Anguis spp. representatives, created with 765 bp of ND2 gene alignment. The tree was created using GTR + I +G model of substitution, as suggested by jModelTest 2.1.10 (Guindon & Gascuel Citation2003; Darriba et al. Citation2012). Tree was generated with 10,000,000 MCMC generations and 25% of burn-in. The individual used for mitogenome sequencing is marked with a star. A homologous sequence of Pseudopus apodus was used as an outgroup. Genbank accession numbers and Bayesian posterior probabilities of nodes are shown on the tree.
The complete mitochondrial genome of the A. veronensis (in total 17,322 bp; GenBank KX236332) has the same gene order and organization as mitogenomes of A. fragilis and A. cephallonica. Mitochondrial DNA of A. veronensis is shorter than mtDNA of the A. fragilis (17479 bp) but longer than the one of A. cephallonica (17208 bp). Differences in mtDNA length are the result of control region length polymorphism (1779 bp for A. cephallonica, 1891 bp for A. veronensis, and 2033 for A. fragilis). Overall base composition of A. veronensis H-strand is: A – 30.8%, T – 24.9%, C – 29.9%, G – 14.4%. In all three mitogenomes start codons have the same nucleotide composition with the exception of ND3 and ND5 (e.g.: ATG in A. veronensis and A. cephallonica; GTG in A. fragilis). As in A. cephallonica, six protein-coding genes truncated stop codons were identified in A. veronensis mitogenome, whereas only five truncated stop codons are present in A. fragilis. Anguis veronensis mitogenome has the highest A + T bias (55.7%) from all already known slow-worms mitochondrial genomes (54.7% for A. cephallonica and 55.2% for A. fragilis).
Acknowledgements
D. Jab. was supported by the grants VEGA 1/0073/14, UK/20/2014 and UK/37/2015. This work was supported by the Slovak Research and Development Agency under the contract no. APVV-0147-15 and by Wroclaw Centre of Biotechnology, program ‘The Leading National Research Centre (KNOW) of Wroclaw University of Environmental and Life Sciences for years 2014-2018’. AC was supported by an Investigador FCT (IF) grant from the Portuguese ‘Fundaçao para a Ciencia e a Tecnologia’ (IF/00209/2014). We would like to thank to Peter Mikulíček and David Jandzik for cooperation and comments on manuscript.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Additional information
Funding
References
- Albert EM, San Mauro D, García-París M, Rüber L, Zardoya R. 2009. Effect of taxon sampling on recovering the phylogeny of squamate reptiles based on complete mitochondrial genome and nuclear gene sequence data. Gene. 441:12–21.
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69:313–319.
- Darriba D, Taboada GL, Doallo R, Posada D. 2012. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 9:772.
- Douglas D.A, Janke A, Arnason U. 2006. A mitogenomic study on the phylogenetic position of snakes. Zool Scr. 35:545–558.
- Douglas D.A, Gower DJ. 2010. Snake mitochondrial genomes: phylogenetic relationships and implications of extended taxon sampling for interpretations of mitogenomic evolution. BMC Genomics. 11:14.
- Guindon S, Gascuel O. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 52:696–704.
- Gvoždík V, Benkovský N, Crottini A, Bellati A, Moravec J, Romano A, Sacchi R, Jandzik D. 2013. An ancient lineage of slow worms, genus Anguis (Squamata: Anguidae), survived in the Italian Peninsula. Mol Phylogenet Evol. 69:1077–1092.
- Gvoždík V, Jandzik D, Lymberakis P, Jablonski D, Moravec J. 2010. Slow worm, Anguis fragilis (Reptilia: Anguidae) as a species complex: genetic structure reveals deep divergences. Mol Phylogenet Evol. 55:460–472.
- Jablonski D, Jandzik D, Mikulíček P, Džukić G, Ljubisavljević K, Tzankov N, Jelić D, Thanou E, Moravec J, Gvoždík V. 2016. Contrasting evolutionary histories of the legless lizards slow worms (Anguis) shaped by the topography of the Balkan Peninsula. BMC Evol Biol. 16:99.
- Mezzasalma M, Guarino FM, Aprea G, Petraccioli A, Crottini A, Odierna G. 2013. Karyological evidence for diversification of Italian slow worm populations (Squamata, Anguidae). Comp Cytogenet. 7:217–227.
- Nabholz B, Uwimana N, Lartillot N. 2013. Reconstructing the phylogenetic history of long-term effective population size and life-history traits using patterns of amino acid replacement in mitochondrial genomes of mammals and birds. Genome Biol Evol. 5:1273–1290.
- Ronquist F, Teslenko M, Van Der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61:539–542.
- Strzała T, Grochowalska R, Najbar B, Mikulíček P, Jandzik D, Lymberakis P, Jablonski D. 2016. Complete mitochondrial genome of the endemic legless lizard Anguis cephallonica Werner, 1894 and its comparison with mitogenome of Anguis fragilis Linnaeus, 1758. Mitochondrial DNA Resour Part B. 1:1:3.