
Abstract
We sequenced and assembled three whole mitogenome sequences of the commercially important snakeskin gourami Trichopodus pectoralis isolated from Malaysia (introduced), Viet Nam (native) and Thailand (native). The mitogenome length range from 16,397 to 16,420 bp. The final partitioned nucleotide alignment consists of 14,002 bp and supports the monophyly of the genus Trichopodus (95% ultrafast bootstrap support) with T. trichopterus forming a sister group with the members of T. pectoralis.
The snakeskin gourami Trichopodus pectoralis commercially used as ornamental and food fish, and it is the largest and most significant species among the trichogastrids. It is native to the central plain of Thailand, extending from Chao Phraya Basin to Cambodia and Vietnam, Laos (Boonsom Citation1985), but, it is now prevalent across Southeast Asia countries due to numerous past introductions. Three snakeskin gourami specimens were collected from Vietnam, Thailand, and Malaysia, and stored at the Institute of Oceanography, Universiti Malaysia Terengganu, with voucher ID UMTGen01310–01312. Genomic DNA (gDNA) extraction from ∼20 mg of fin clip was performed using the Solokov protocol (Sokolov Citation2000). Approximately 100–200 ng of purified gDNA was subsequently sheared to 500 bp using a M220 focused-ultrasonicator (Covaris, Woburn, MA), processed using NEBNext Ultra DNA library prep kit and sequenced on the MiSeq desktop sequencer (Illumina, San Diego, CA) located at the Monash University Malaysia Genomics Facility using a run configuration of 2 × 250 bp. For isolates UMTGen01311 and UMTGen01312, mitogenome assembly was performed using MITObim v 1.8 (Hahn et al. Citation2013), using the COX1 gene fragment of the species as the bait for iterative mapping assembly. For isolate UMTGen01311, the complete mitogenome of isolate UMTGen01312 was used instead of the bait for reference-based assembly. Each mitogenome was circularized (Gan et al. Citation2014) and annotated using MitoAnnotator (Iwasaki et al. Citation2013).
Protein coding genes (PCGs) were partitioned and aligned based on gene and codon position using TranslatorX (Abascal et al. Citation2010) while the 12S and 16S ribosomal RNA genes aligned using Muscle, generating 41 nucleotide alignments (13 PCGs x 3 codon positions +2 rRNA). Each alignment was subsequently trimmed with TrimAl version 1.9 (Capella-Gutiérrez et al. Citation2009) and concatenated. Best-fit partitioning scheme was calculated using ModelFinder version 1.5.3 (Nguyen et al. Citation2015) and a maximum-likelihood (ML) tree was constructed with 1000 ultrafast bootstrap approximation option. The consensus tree was visualized using FigTree 1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/). Additional nucleotide similarity search to verify the taxonomy of Trichopodus microlepis (NC_027238) was performed using BlastN (Altschul et al. Citation1997).
ModelFinder identified five partitions as the best-fit partitioning scheme as follows (-# after gene name indicates codon position): Partition 1 = 12S and 16S rRNA; Partition2 = ATP6-1,NAD3-1,NAD4-1,COB-1,NAD4L-1,NAD1-1,NAD2-1,ATP8-1, NAD5-1,COX1-1,COX2-1,COX3-1,NAD6-1; Partition3 = ATP6-2, NAD4-2,NAD6-2,NAD2-2,ATP8-2,NAD5-2,COB-2,NAD4L-2,NAD1-2, NAD3-2,COX2-2,COX1-2,COX3-2; Partition 4 = ATP6-3,NAD1-3,NAD3-3,NAD5-3,NAD4-3,COX3-3,NAD4L-3,COB-3,ATP8-3,COX2-3, NAD2-3,COX1-3 and Partition 5 = NAD6-3.
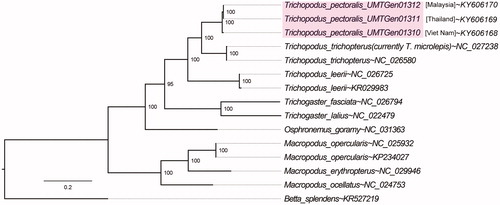
The final partitioned nucleotide alignment consists of 14,002 bp length and supports the monophyly of the genus Trichopodus (95% ultrafast bootstrap support) with Trichopodus trichopterus forming a sister group with members of Trichopodus pectoralis reported in this study (). It is also worth noting that GenBank entry NC_027238 was initially submitted as Trichopodus microlepis, however the COX1, COB, and 12S rRNA genes of this mitogenome was consistently lower than 90% identity to similar gene fragments from vouchered specimens in NCBI database (Accession codes: KU569058, KU569059, KF805359, KF805360, AY763711 and AY763757), suggesting potential misidentification and the need to sequence the whole mitogenome of a correctly identified Trichopodus microlepis specimen.
Figure 1. Evolutionary relationships of Trichopodus pectoralis reported in this study (shaded) as inferred from maximum-likelihood estimation based on 13 mitochondrial protein-coding genes and 2 ribosomal RNA genes with best-fit partitioning scheme with Betta splendens rooted as the outgroup. NCBI accession numbers are provided next to tilde (∼) symbols. Numbers at nodes indicate Ultrafast Bootstrap support and branch lengths indicate the number of substitutions per site.
Acknowledgements
The authors would like to thank Yin Peng Lee (Monash University Malaysia) for her assistance in DNA extraction and MiSeq sequencing.
Disclosure statement
The authors report no conflicts of interest and are responsible for the content and writing of the manuscript.
Additional information
Funding
Related Research Data
References
- Abascal F, Zardoya R, Telford MJ. 2010. TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 38:W7–13.
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402.
- Boonsom J. (1985). Aquaculture practices, planning and extension in Thailand. Bangkok: FAO. Chapter 12, Pla salid (Trichogaster pectoralis Regan): a life history and manual for culture; [cited 2017 Feb 14]. Available from: http://www.fao.org/docrep/field/003/ac231e/AC231E12.htm
- Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 25:1972–1973.
- Gan HM, Schultz MB, Austin CM. 2014. Integrated shotgun sequencing and bioinformatics pipeline allows ultra-fast mitogenome recovery and confirms substantial gene rearrangements in Australian freshwater crayfishes. BMC Evol Biol. 14:19.
- Hahn C, Bachmann L, Chevreux B. 2013. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads—a baiting and iterative mapping approach. Nucleic Acids Res. 41:e129–e129.
- Iwasaki W, Fukunaga T, Isagozawa R, Yamada K, Maeda Y, Satoh TP, Sado T, Mabuchi K, Takeshima H, Miya M, Nishida M. 2013. MitoFish and MitoAnnotator: a mitochondrial genome database of fish with an accurate and automatic annotation pipeline. Mol Biol Evol. 30:2531–2540.
- Nguyen LT, Schmidt HA, Von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32:268–274.
- Sokolov EP. 2000. An improved method for DNA isolation from mucopolysaccharide-rich molluscan tissues. J Molluscan Stud. 66:573–575.