
Abstract
We assembled the mitogenome of Cypseloides fumigatus based on off-target sequences from ultraconserved elements sequencing. We found a total length of 16,850 bp, including 13 protein-coding genes, 22 tRNA genes, 2 rRNA genes, and one control region, organized in the standard avian gene order. We have built a phylogenetic tree including 26 species of swifts that suggested C. fumigatus as sister species of C. cryptus, and indicated exciting opportunities for biogeographic inferences involving most continents, including Neartic vs Neotropical disjunctions and local radiations across the globe. Finally, we found cases of lack of reciprocal monophyly between named species and high intra-specific divergence, suggesting that population-level studies are warranted.
Swifts and swiftlets (family Apodidae) comprise a globally distributed group of small to medium-sized insectivorous birds closely related to hummingbirds. Examples of interesting aspects of their biology include their high-speed flight, large portion of their lifetime spent in flight and little variation in plumage colouration (Chantler & Driessens Citation2000). Here we present the complete mitogenome of the Sooty Swift (Cypseloides fumigatus), a relatively rare species of the Cypseloidinae subfamily distributed in Argentina, Bolivia, Brazil, and Paraguay. This is the third swift genome sequenced to date, the other being Apus apus (Morgan-Richards et al. Citation2008, NC_008540.1) and Chaetura pelagica (Xu & Zhang Citation2015, NC_028545.1). We obtained genomic DNA from a muscle sample of an individual collected at Ortigueira, PR, Brazil (24°12′S 50°55′ W, deposited at LGEMA USP tissue collection under #11411), using the Qiagen DNeasy kit (Valencia, CA) with an RNAse treatment. We obtained the mitogenome (Genbank KY688216) from off-target sequences generated during next-generation sequencing of ultraconserved elements (UCEs, performed at Rapid Genomics LLC, Gainesvile, FL), as described in Amaral et al. (Citation2015). We manually aligned four fragments resulting from the mtDNA mining protocol, whose lengths varied from 14,923 bp to 16,872 bp., in Bioedit (Hall Citation1999). We obtained a final consensus sequence of 33,722 bp, which we used as input for automatic annotation in DOGMA (Wyman et al. Citation2004; http://dogma.ccbb.utexas.edu) and MITOS (Bernt et al. Citation2013; http://mitos.bioinf.uni-leipzig.de/index.py) using default parameters. We manually adjusted the automatic annotations using the mitogenome of Apus apus as reference to resolve discordances between the two automatic methods.
We obtained an annotated mitogenome of 16,850 bp, 187 bp shorter than the genome of Apus apus, and that included 2 rRNAs, 22 tRNAs, 13 protein-coding genes, and 1 control region. Gene order was the same found in Apus apus, which is identical to one thought to be ancestral among birds (Gibb et al. Citation2007). Base composition (A = 31.0%, C = 30.6%, G = 13.6%, and T = 24.8%) was similar to that reported for Apus apus. Interestingly, NADH dehydrogenase subunit 3 in C. fumigatus had an extra untranslated base, which has been reported in a number of bird species (Mindell et al. Citation1998) but is absent in the two other published swift mitogenomes. The control region has a long stretch of repetitive sequences precluded establishing the exact number of repetitions, and thus the exact length of the mitogenome may vary slightly from one reported here.
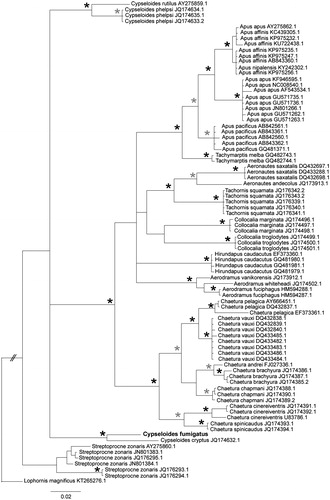
We perfomed a phylogenetic analysis including 622 bp of COI from the sequence obtained here and other 25 Apodidae species available at Genbank, totalling 83 sequences. We aligned the sequences using ClustalW (Larkin et al. Citation2007) as implemented in Geneious 9.1.6 (Kearse et al. Citation2012). We carried out model selection using Akaike information criterion in MrModeltest 2 (Nylander Citation2004). We performed a Bayesian phylogenetic inference in MrBayes 3.2.6 (Huelsenbeck & Ronquist Citation2001) using GTR + I + G, 10.000.000 generations, sampling frequency of 5000, burnin of 25% and Lophornis magnificus as an outgroup. The phylogenetic analysis indicated that C. fumigatus and C. cryptus are sister species (), suggesting a split between eastern and western Neotropics. Despite limited resolution, our tree also suggested interesting historical events as Neotropical vs Neartic disjunctions (e.g. clade Tachornis/Aeronautes), local radiations entirely or mostly restricted to the Old World (e.g. Apus), Neotropics (Chaetura) and Asia, Oceania and Australia (Aerodramus). In addition, we found high intra-specific divergences in S. zonaris, C. cinereiventris and haplotype sharing among Apus species, what indicate exciting opportunities for population-level studies.
Figure 1. Bayesian phylogenetic inference based on COI sequences of swifts. Accession numbers are indicated at the right of species names. Stars correspond to posterior probabilities of 1.0 (black), or equal to or within the interval of 0.95 to 0.99 (gray). Posterior probabilities lower than 0.95 were not indicated.
Acknowledgements
We thank Renato Gaban Lima for collecting the specimen used here, and Cristina Y. Miyaki for loaning the tissue sample under her care at LGEMA (Laboratório de Genética e Evolução Molecular de Aves, USP).
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Additional information
Funding
References
- Amaral FR, Neves LG, Resende Jr MFR, Mobili F, Miyaki CY, Pellegrino KCM, Biondo C. 2015. Ultraconserved elements sequencing as a low-cost source of complete mitochondrial genomes and microsatellite markers in non-model amniotes. PLoS One. 10:e0138446.
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69:313–319.
- Chantler P, Driessens G. 2000. Swifts: a guide to the swifts and treeswifts of the world. New Haven: Yale University Press.
- Gibb GC, Kardailsky O, Kimball RT, Braun EL, Penny D. 2007. Mitochondrial genomes and avian phylogeny: complex characters and resolvability without explosive radiations. Mol Biol Evol. 24:269–280.
- Hall TA. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Series. 41:95–98.
- Huelsenbeck JP, Ronquist F. 2001. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 17:754–755.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28:1647–1649.
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al. 2007. Clustal W and Clustal X version 2.0. Bioinformatics. 23:2947–2948.
- Mindell DP, Sorenson MD, Dimcheff DE. 1998. An extra nucleotide is not translated in mitochondrial ND3 of some birds and turtles. Mol Biol Evol. 15:1568–1571.
- Morgan-Richards M, Trewick SA, Bartosch-Härlid A, Kardailsky O, Phillips MJ, McLenachan PA, Penny D. 2008. Bird evolution: testing the Metaves clade with six new mitochondrial genomes. BMC Evolut Biol. 8:20.
- Nylander JAA. 2004. MrModeltest v2. Program distributed by the author. Evolutionary Biology Centre, Uppsala University.
- Xu XQ, Zhang K. 2015. Complete mitochondrial genome and phylogenetic analysis of the Chimney Swift, Chaetura pelagica. Mitochondrial DNA. 29:1–2.
- Wyman SK, Jansen RK, Boore JL. 2004. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 20:3252–3255.