
Abstract
The beluga whale is one of three endemic Arctic whales. The species is philopatric, and its migration patterns are passed from mother to calf. Management of the species is informed by the levels of genetic structuring among summer aggregation sites based on mitochondrial D-Loop data. To assess the levels of differentiation across the entire mitochondrial genome within belugas, we present a comparison between the first two complete mitochondrial genomes from opposite sides of their distribution range: Baffin Bay and the Russian Far East. Our analyses reveal that additional phylogenetic insights can be gained from expanding the genetic region analyzed. Further, we estimate the divergence time between the two mitochondrial genomes to be 0.469 MYA.
Here, we present the complete mitochondrial genome of a beluga whale (white whale, Delphinapterus leucas) from Baffin Bay. The beluga whale is a toothed whale belonging to the family Monodontidae, which also includes the narwhal Monodon monoceros. It has a discontinuous circumpolar distribution and is endemic to the Arctic region. Individuals can be up to 6.7 m long and are easily recognized by their white skin (Stewart & Stewart Citation1989). Mitochondrial DNA sequence information is of particular importance in the conservation of the beluga whale; the species is philopatric and does not re-colonize summer aggregation sites from which it has been extirpated (Brown Gladden et al. Citation1997). Previous studies have reported higher levels of genetic differentiation among summer aggregation sites based on mitochondrial D-Loop data than on up to 15 nuclear microsatellite markers (De March & Postma Citation2003). In light of this, other mitochondrial regions could be used to augment the 400–600 base pairs currently used for management purposes (O’Corry-Crowe et al. Citation2002; Turgeon et al. Citation2012; Meschersky et al. Citation2013).
A beluga whale reference mitochondrial genome from the Sea of Okhotsk, Russia, was recently published (Kim et al. Citation2017). The mitochondrial genome presented here is from Baffin Bay, and therefore represents the opposite end of the distribution range of the species. The analysis of this specimen may identify mitochondrial genomic regions of interest for further studies. The individual was sampled during the Inuit subsidence hunt in Qeqertarsuaq (69.237835, −53.526519) in western Greenland in April 2008 by staff from Greenland Institute of Natural Resources. The tissue sample is stored at the Natural History Museum of Denmark (ID number CGG_1_017647). The sequence is available from GenBank under accession number KY888944.
We extracted DNA from skin tissue using the Kingfisher Duo extraction robot and Cell and Tissue DNA Kit from ThermoFisher Scientific using the manufacturer’s protocol (ThermoFisher Scientific, Waltham, MA). Paired-end sequencing was performed on 180 base pair inserts using the Illumina HiSeq X platform (San Diego, CA). We assembled the mitogenome using a combination of MIRA 4.0.2 (Chevreux et al. Citation1999) and MITObim v.1.8 (Hahn et al. Citation2013), using the narwhal reference genome as a reference (Genbank accession: NC005279). We performed the annotation using the MITOS web service (Bernt et al. Citation2013) using default parameters. We performed a phylogenetic analysis using the 13 protein-coding regions across six closely-related toothed whale species and the beluga reference mitochondrial genome (Kim et al. Citation2017). The phylogenetic tree was constructed from the best tree among 100 independent runs in RAxML v.8.2 (Stamatakis Citation2014) using a GTR-GAMMA substitution model (). Topology was verified by 100 bootstraps. The number of variable sites in each genomic region was calculated using FaBox 1.4.1 (Villesen Citation2007). Divergence time (T) between the two mitochondrial genomes was calculated as T = K/(2υ), using a mutation rate (υ) estimated for killer whales of 2.60 × 10−3 substitutions per site per million years (1.50–3.83 × 10−3) (Morin et al. Citation2008) and a pairwise differentiation (K) of 2.44 × 10−3 calculated as the number of variable sites per site.
Figure 1. Phylogenetic tree of beluga whales from Baffin Bay and Sea of Okhotsk, and five related toothed whale species.
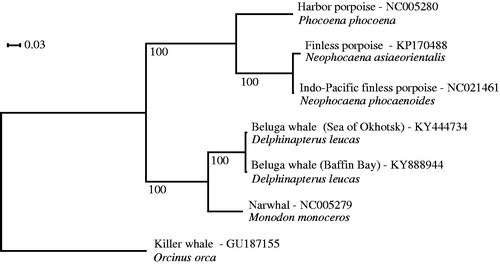
Our assembly yielded a 16,386 base-pair circular genome with gene regions in accordance with the findings of Kim et al. (Citation2017). We found a total of 40 variable sites between the two beluga mitochondrial genomes, with 4, 2, 6 and 28 variable sites in the tRNA, rRNA, D-loop and protein-coding regions, respectively. The rRNAs had one variable site each and the protein-coding regions had between 1 and 5 variable sites (ND1:1, ND2:5, COX1:3, COX2:2, ATP8:1, ATP6:1, COX3:2, ND3:1 ND4L:1, ND4:2, ND5:4, ND6:3, CYTB:2), indicating that additional phylogenetic information is available in the mitochondrial genome relative to the D-Loop. We estimated a divergence time between the two mitochondrial linages of 0.469 (0.319–0.814) MYA, which is the same order of magnitude as the divergence time estimation of killer whale lineages (Morin et al. Citation2008).
Acknowledgements
The authors acknowledge support from Science for Life Laboratory, the Knut and Alice Wallenberg Foundation, the National Genomics Infrastructure funded by the Swedish Research Council, and Uppsala Multidisciplinary Center for Advanced Computational Science for assistance with massively parallel sequencing and access to the UPPMAX computational infrastructure.
Disclosure statement
The authors report no conflicts of interest and are alone responsible for the content and writing of the paper.
Additional information
Funding
References
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69:313–319.
- Brown Gladden JG, Ferguson MM, Clayton JW. 1997. Matriarchal genetic population structure of North American beluga whales Delphinapterus leucas (Cetacea: Monodontidae). Mol Ecol. 6:1033–1046.
- Chevreux B, Wetter T, Suhai S. (1999). Genome sequence assembly using trace signals and additional sequence information. Comp Sci Bio. Proceedings of the German Conference on Bioinformatics (GCB). 45–56.
- De March BGE, Postma LD. 2003. Molecular genetic stock discrimination of belugas (Delphinapterus leucas) hunted in eastern Hudson Bay, northern Quebec, Hudson Strait, and Sanikiluaq (Belcher Islands), Canada, and comparisons to adjacent populations. Arctic. 56:111–124.
- Hahn C, Bachmann L, Chevreux B. 2013. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads – a baiting and iterative mapping approach. Nucleic Acids Res. 41:e129.
- Kim JH, Lee YR, Koh JR, Jun JW, Giri SS, Kim HJ, Chi C, Yun S, Kim SG, Kim SW, et al. 2017. Complete mitochondrial genome of the beluga whale Delphinapterus leucas (Cetacea: Monodontidae). Conserv Genet Res. 1–4.
- Meschersky IG, Shpak OV, Litovka DI, Glazov DM, Borisova EA, Rozhnov VV. 2013. A genetic analysis of the beluga whale Delphinapterus leucas (Cetacea: Monodontidae) from summer aggregations in the Russian Far East. Russ J Mar Biol. 39:125–135.
- Morin PA, Archer FI, Foote AD, Vilstrup J, Allen EE, Wade P, Durban J, Parsons K, Pitman R, Li L, et al. 2008. Complete mitochondrial genome phylogeographic analysis of killer whales (Orcinus orca) indicates multiple species. Genome Res. 20:908–916.
- O’Corry-Crowe GM, Dizon AE, Suydam RS, Lowry LF. (2002). Molecular genetic studies of population structure and movement patterns in a migratory species: the beluga whale, Delphinaptevus leucas, in the western Nearctic. In: Pfeiffer CJ, editor. Molecular and cell biology of marine mammals. Malabar, FL: Krieger Publishing Company; p. 56–64.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30:1312–1313.
- Stewart BE, Stewart REA. 1989. Delphinapterus leucas. Mamm Species. 336:1–8.
- Turgeon J, Duchesne P, Colbeck GJ, Postma LD, Hammill MO. 2012. Spatiotemporal segregation among summer stocks of beluga (Delphinapterus leucas) despite nuclear gene flow: implication for the endangered belugas in eastern Hudson Bay (Canada). Conserv Genet. 13:419–433.
- Villesen P. 2007. FaBox: an online toolbox for FASTA sequences. Mol Ecol Notes. 7:965–968.