
Abstract
The complete mitochondrial genomes are sequenced in two individuals representing two morphological forms, ‘usual’ (U) and ‘gray’ (G), of the short-spined sea urchin Strongylocentrotus intermedius. The genome sequences are 15,705 bp in size, and the gene arrangement, composition, and size are very similar to the other sea urchin mitochondrial genomes published previously. A low level of sequence divergence (Dxy = 0.0083 ± 0.0007) is detected between the forms. The GenBank (KC490912) mt genome of S. intermedius is much closer to the U form (Dxy = 0.0013 ± 0.0003) than to the G form (Dxy = 0.0085 ± 0.0006), demonstrating unique evolutionary trajectories for each form, which we previously suggested based on the bindin gene and symbiont analyses.
The short-spined sea urchin Strongylocentrotus intermedius (A. Agassiz) inhabits a wide range of the northwest Pacific region, including the Sea of Japan and the Sea of Okhotsk (Jensen Citation1974; Bazhin and Stepanov Citation2012). There are two sympatric morphological forms, ‘usual’ (U) and ‘gray’ (G), which are different in morphology and preferred bathymetric distribution. We detected previously that these two forms harbour highly divergent bacterial symbiont lineages and show distinct patterns of nuclear bindin gene variability but not mitochondrial COI gene (Balakirev et al. Citation2008, Citation2016). The evolutionary history and taxonomical relationships of the S. intermedius morphological forms remain unclear due to limited genetic data.
We have sequenced two complete mitochondrial (mt) genomes of S. intermedius represented by two morphological forms (GenBank accession numbers KY964299, KY964300) from a single sea urchin settlement within a distance of ∼150 m (46°15.086′ N, 138°06.646′ E; Cape Zolotoi, Sea of Japan). The U and G forms were collected at depths of 5–10 m and 15–25 m, respectively. The mt fragments were amplified with primers designed with the program mitoPrimer, v. 1 (Yang et al. Citation2011). The tissue samples are stored at the A. V. Zhirmunsky Institute of Marine Biology museum (Vladivostok, Russia); accession numbers MIMB 33945 and MIMB 33946.
The genome sequences of S. intermedius are 15,705 bp in size, and the gene arrangement, composition, and size are very similar to other strongylocentrotid sea urchin mitochondrial genomes published previously. There are 131 single nucleotide and one length (within the ND6 gene) differences between the two haplotypes (SIT17 and SIG19); total sequence divergence (Dxy) is 0.0083 ± 0.0007. Comparison of the new mt genomes with mt genomes available in GenBank for the genera Strongylocentrotus, Mesocentrotus, and Pseudocentrotus reveals a close affinity of S. intermedius to other strongylocentrotid sea urchins () with low divergence (Dxy = 0.0049 ± 0.0004) between our specimens and the GenBank S. intermedius (KC490912) complete mt genome.
Figure 1. Maximum likelihood tree for the short-spined sea urchin Strongylocentrotus intermedius specimens SIG19 and SIT17, and GenBank representatives of the class Echinoidea. The tree is constructed using whole mitochondrial genomes. The tree is based on the general time reversible + gamma + invariant sites (GTR + G + I) model of nucleotide substitution. The numbers at the nodes are bootstrap percent probability values based on 500 replications (values below 75% are omitted).
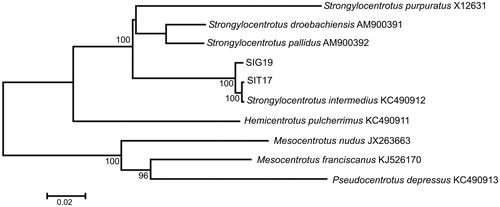
The GenBank S. intermedius specimen (KC490912) was collected from a depth less than 10 m in Jumunjin Harbor, Sea of Japan, South Korea (Dr. Youn-Ho Lee, Korea Institute of Ocean Science and Technology) and represented the U form inhabiting shallow-water settlements (5–10 m). The Jumunjin Harbor is ∼2089.43 km along the coastline from Cape Zolotoi, where we collected our specimens. Despite the long distance, the nucleotide difference between the GenBank and the U form is low (Dxy = 0.0013 ± 0.0003), but 6.5 times higher (Dxy = 0.0085 ± 0.0006) between the GenBank specimen and the G form. Thus, our specimens, collected in close proximity (∼150 m) but belonging to different morphological forms, are more different than specimens collected from distant regions (separated by ∼2089.43 km) but belonging to the same morphological form. These discrepancies between genetic differences and geographical distances confirm that the U and G morphological forms of S. intermedius represent distinct evolutionary lineages (incipient species) with low genetic divergence but significant differences in symbiont contents (Balakirev et al. Citation2008, Citation2016).
Acknowledgements
We appreciate Dr. G. M. Kamenev, N. V. Kameneva, and I. A. Dyachenko (National Scientific Center of Marine Biology, Vladivostok, Russia) for estimating the exact geographical coordinates of the Cape Zolotoi (the Sea of Japan). The research on mitochondrial genome sequencing was conducted at the Department of Ecology and Evolutionary Biology, University of California, Irvine, USA. The data analysis and manuscript preparation were conducted at the A. V. Zhirmunsky Institute of Marine Biology, Vladivostok, Russia.
Disclosure statement
The funders had no role in the study’s design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors alone are responsible for the content and writing of the paper. The authors acknowledge no financial interest or benefit. The authors report that they have no conflicts of interest.
Additional information
Funding
References
- Balakirev ES, Pavlyuchkov VA, Anisimova M, Ayala FJ. 2016. DNA polymorphism and selection at the bindin locus in three Strongylocentrotus sp. (Echinoidea). BMC Genet. 17:66.
- Balakirev ES, Pavlyuchkov VA, Ayala FJ. 2008. DNA variation and symbiotic associations in phenotypically diverse sea urchin Strongylocentrotus intermedius. Proc Natl Acad Sci USA. 105:16218–16223.
- Bazhin AG, Stepanov VG. 2012. In: Levin VS, editor. Sea urchins fam. Strongylocentrotidae of seas of Russia. Petropavlovsk-Kamchatsky: KamchatNIRO; 196 p.
- Jensen M. 1974. The Strongylocentrotidae (Echinoidea), a morphologic and systematic study. Sarsia. 57:113–148.
- Yang CH, Chang HW, Ho CH, Chou YC, Chuang LY. 2011. Conserved PCR primer set designing for closely-related species to complete mitochondrial genome sequencing using a sliding window-based PSO algorithm. PLoS ONE. 6:e17729.