
Abstract
In this study, the complete 17,809 bp mitochondrial genome of Callosobruchus maculates (F.) (Coleoptera: Chrysomelidae: Bruchinae) was sequenced using Illumina’s HiSeq2000 platform. The mitogenome is a double-stranded circular molecule of 17,809 bp in length with 21 transfer RNA genes, 13 protein-coding genes, and two ribosomal RNA genes as in other insects. Specially, there is a 2008 bp-inserted segment between ND2 and tRNA-Trp from 1180 to 3187, which cannot be aligned to any known gene of mitogenomes. To estimate the taxonomic status of Bruchinae, total 17 species from eight subfamilies of Chrysomelidae were selected as ingroups and three species of Lamiinae as outgroups for phylogenetic analysis based on mitogenome. The results showed that three major lineages were formed, including a basal ‘Eumolpine’ clade (Cassidinae, Eumolpinae, Cryptocephalinae, Clytrinae), ‘'Criocerine’ clade (Criocerinae, Bruchinae) and ‘Chrysomeline’ clade (Chrysomelinae, Galerucinae s. l.). Bruchinae showed more closed relationship with Criocerinae than other subfamilies. More thorough taxon sampling will be needed to well understand the relationship in Chrysomelidae.
Callosobruchus maculates (F.) (Coleoptera: Chrysomelidae: Bruchinae) is an important insect pest of cowpea in the store causing considerable damage to the grains. During larval stages, it causes substantial quantitative and qualitative losses (50–90%) manifested by seed perforation and reductions in weight, mark value, and germination ability of seeds (Ofuya and Osadahun Citation2005; Brisibe et al. Citation2011; Akami et al. Citation2016). The beetle most likely originated in West Africa and moved around the globe with the trade of legumes and other crops (Tran and Credland Citation1995). Now, this pest of stored legumes has a cosmopolitan distribution.
Traditionally, bean weevil was considered a separate family within Chrysomelidae (Crowson Citation1955; Kingsolver Citation1996; Reid Citation1996; Verma and Saxena Citation1996; Duckett Citation1997; Lingafelter and Pakaluk Citation1997; Verma Citation1998), but the group was demoted to subfamily rank in several phylogenetic studies based on molecular data (Reid Citation1995, Citation2000; Farrell and Sequeira Citation2004; Gómez-Zurita et al. Citation2008; Bocak et al. Citation2014; Haddad and Mckenna Citation2016). According to Bouchard et al. (Citation2011), Chrysomelidae contains 13 subfamilies: Sagrinae, Bruchinae, Donaciinae, Criocerinae, Cassidinae, Chrysomelinae, Galerucinae, Lamprosomatinae, Cryptocephalinae, Eumolpinae, Spilopyrinae, Synetinae, and Protoscelidinae. Recently, with the development of next-generation sequencing, a large-scale mitogenome has been widely used to resolve the phylogeny and evolution of organisms across the tree of Coleoptera (Timmermans et al. Citation2010; Crampton-Platt et al. Citation2015; Gomez-Rodriguez et al. Citation2015; Timmermans et al. Citation2016; Haddad and Mckenna Citation2016; Nie et al. Citation2017). In this study, the complete mitogenome of C. maculates was sequenced by next-generation sequencing technology to estimate the taxonomic status of Bruchinae.
The specimens used in this study were intercepted in imported Vigna unguiculata from Nigeria and deposited in the plant laboratory of Beijing Inspection and Quarantine Testing Center. Genomic DNA was extracted by TIANprep Midi Plasmid kit (TIANGEN, Beijing, China) and then sequenced using Illumina’s HiSeq2000 platform (Illumina, San Diego, CA) with 200 bp insert size and a pair-end 100 bp sequencing strategy. The sequence reads were first filtered by the programs following Zhou et al. (Citation2013) and then the remaining high-quality reads were assembled using SOAPdenovo-Trans (Xie et al. Citation2014). The annotations of genes were done by Geneious 8.0.5 software (Kearse et al. Citation2012) and tRNAscan-SE 1.21 (Schattner et al. Citation2005). In order to confirm the insert gene between ND2 and tRNA-Trp from 1180 to 3187, we sequenced this species twice sampling two specimens using Illumina’s HiSeq2000 platform and Illumina’s HiSeq2500 platform.
The complete mitochondrial genome (mitogenome) of C. maculates is a double-stranded circular molecule of 17,809 bp in length (GenBank accession number: MF960125), with 21 transfer RNA genes (tRNA-Gln lost), 13 protein-coding genes, and two ribosomal RNA genes as in other insects. The overall base composition is A: 45.5%, T: 34.8%, C: 13.6%, and G: 6.1%, with a much higher A + T content. Specially, there is 2008 bp inserted genes between ND2 and tRNA-Trp from 1180 to 3187, which cannot be aligned to any known gene of mitogenome. We had blasted and tried to annotate the insert gene. However, there was no gene matching this fragment. It will be worthy to add more mitogenomes of other species of Bruchinae to explore the potential function of inserted genes and why the mitogenome lost tRNA-Gln.
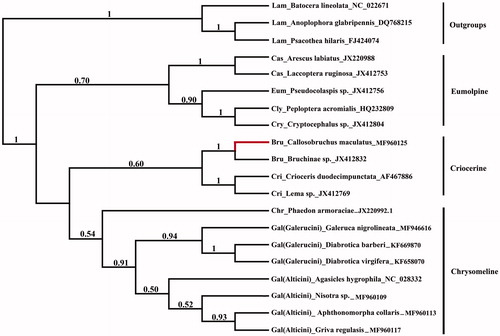
The phylogenetic tree was reconstructed to estimate the status of Bruchinae in Chrysomelidae. All available mitogenomes of subfamilies of Chrysomelidae were downloaded from Genbank. The acceptable sequences including 13 protein-coding genes and longer than 10K bp were kept. Total 17 species from eight subfamilies (accession numbers: JX220988, JX412753, JX412756, HQ232809, JX412804, MF960125, JX412832, AF467886, JX412769, JX220992.1, MF946616, KF669870, KF658070, NC_028332, MF960109, MF960113, MF960117) were selected as ingroups and three species of Lamiinae (accession numbers: DQ768215, NC_022671, FJ424074) was selected as outgroups. The combined data set of 13 protein-coding gene (PCGs) were aligned with TransAlign (Bininda-Emonds Citation2005). The data aligned from 13PCGs were concatenated with Sequence Matrix v.1.7.8 (Vaidya et al. Citation2011). The Bayesian phylogenetic inference was performed using MrBayes v.3.2 (Ronquist et al. Citation2012) based on the combined data set of 13 PCGs. Data were partitioned according to loci of 13 PCGs. The MCMC search was conducted for 1,000,000 generations, and sampling was done every 100 generations until the average standard deviation of split frequencies was below 0.01. The first 25% of trees were discarded as ‘burn-in’ and posterior probabilities were estimated for each node.
Phylogenetic analyses () showed that three major lineages were formed, including a basal ‘Eumolpine’ clade (Cassidinae, Eumolpinae, Cryptocephalinae, Clytrinae), ‘Criocerine’ clade (Criocerinae, Bruchinae), and ‘Chrysomeline’ clade (Chrysomelinae, Galerucinae s. l.). Bruchinae showed more closed relationship with Criocerinae than other subfamilies. However, more thorough taxon sampling will be needed to well understand the relationship in Chrysomelidae because mitogenomes of only eight subfamilies were involved in the present study.
Figure 1. The Bayesian tree based on 13 PCGs combined data sets. Numbers on nodes indicate Bayesian posterior probabilities. Gray branch is the new data in this study.
Acknowledgments
We thank Min Tang and Xin Zhou who are from China National GeneBank-Shenzhen, BGI-Shenzhen, Yantian District, Shenzhen, China for help with sequence.
Disclosure statement
The authors have declared no competing interests.
Additional information
Funding
Related Research Data
References
- Akami M, Niu C, Chakira H, Chen Z, Vi T, Nukenine EN. 2016. Persistence and comparative pesticidal potentials of some constituents of Lippia adoensis (Hochst. ex Walp.) (Lamiales: Verbenaceae) essential oil against three life stages of Callosobruchus maculatus (Fab.) (Coleoptera: Bruchidae). Brit Biotech J. 13:1–16.
- Bininda-Emonds O. 2005. TransAlign: using amino acids to facilitate the multiple alignment of protein-coding DNA sequences. BMC Bioinf. 6:156.
- Bocak L, Barton C, Crampton-Platt A, Chesters D, Ahrens D, Vogler AP. 2014. Building the Coleoptera tree-of-life for >8000 species: composition of public DNA data and fit with Linnaean classification. Syst Entomol. 39:97–110.
- Bouchard P, Bousquet Y, Davies AE, Alonso-Zarazaga MA, Lawrence JF, Lyal CHC, Newton AF, Reid CAM, Schmitt M, Ślipiñski SA, et al. 2011. Family-group names in Coleoptera (Insecta). ZooKeys. 88:1–972.
- Brisibe EA, Adugbo SE, Ekanem U, Brisibe F, Figueira GM. 2011. Controlling bruchid pests of stored cowpea seeds with dried leaves of Artemisia annua and two other common botanicals. Afr J Biotechnol. 10:9586–9592.
- Crampton-Platt A, Timmermans MJ, Gimmel ML, Kutty SN, Cockerill TD, Khen CV, Vogler AP. 2015. Soup to tree: the phylogeny of beetles inferred by mitochondrial metagenomics of a Bornean rainforest sample. Mol Biol Evol. 32:2302–2316.
- Crowson RA. 1955. The natural classification of the families of Coleoptera. London: Nathaniel Lloyd.
- Duckett CN. 1997. The scientific method and the predictive value of classification. Chrysomela. 34:3–4.
- Farrell BD, Sequeira AS. 2004. Evolutionary rates in the adaptive radiation of beetles on plants. Evolution. 58:1984–2001.
- Gomez-Rodriguez C, Crampton-Platt A, Timmermans MJ, Baselga A, Vogler AP. 2015. Validating the power of mitochondrial metagenomics for community ecology and phylogenetics of complex assemblages. Meth Ecol Evol. 6:883–894.
- Gómez-Zurita J, Hunt T, Vogler AP. 2008. Multilocus ribosomal RNA phylogeny of the leaf beetles (Chrysomelidae). Cladistics. 24:34–50.
- Haddad S, Mckenna DD. 2016. Phylogeny and evolution of the superfamily Chrysomelidae (Coleoptera: Cucujiformia). Syst Entomol. 41:697–716.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28:1647–1649.
- Kingsolver JM. 1996. On the family Bruchidae. Chrysomela. 30:3.
- Lingafelter S, Pakaluk J. 1997. Comments on the bruchine Chrysomelidae. Chrysomela. 33:3.
- Nie RE, Breeschoten T, Timmermans MJTN, Nadein K, Xue HJ, Bai M, Huang Y, Yang XK, Vogler AP. 2017. The phylogeny of Galerucinae (Coleoptera: Chrysomelidae) and the performance of mitochondrial genomes in phylogenetic inference compared to nuclear rRNA genes. Cladistics. 33:1–18.
- Ofuya TL, Osadahun JM. 2005. Effect of three plant powders on behaviour, mortality and reproductive fitness of Callosobruchus maculatus (Fabricius) (Coleoptera: Bruchidae). Zool Sci. 26:603–608.
- Reid CAM. 1995. A cladistic analysis of subfamilial relationships in the Chrysomelidae sensu lato (Chrysomeloidea). In: Pakaluk J, Slipinski SA, editors. Biology, phylogeny, and classification of Coleoptera: papers celebrating the 80th birthday of Roy A. Crowson. Vol. 2. Warszawa: Muzeum i Instytut Zoologii PAN; p. 559–631.
- Reid CAM. 1996. More on the family Bruchidae. Chrysomela. 31:3.
- Reid CAM. 2000. Spilopyrinae Chapuis: a new subfamily in the Chrysomelidae and its systematic placement (Coleoptera). Invertebr Taxon. 14:837–862.
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61:539–542.
- Schattner P, Brooks AN, Lowe TM. 2005. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33:686–689.
- Timmermans MJTN, Barton C, Haran J, Ahrens D, Culverwell CL, Ollikainen A, Dodsworth S, Foster PG, Bocak L, Vogler AP. 2016. Family-level sampling of mitochondrial genomes in Coleoptera: compositional heterogeneity and phylogenetics. Genome Biol Evol. 8:161–175.
- Timmermans MJTN, Dodsworth S, Culverwell C, Bocak L, Ahrens D, Littlewood D, Pons J, Vogler AP. 2010. Why barcode? High-throughput multiplex sequencing of mitochondrial genomes for molecular systematics. Nucleic Acids Res. 38:e197.
- Tran BMD, Credland PF. 1995. Consequences of inbreeding for the cowpea seed beetle, Callosobruchus maculates (F.) (Coleoptera: Bruchidae). Biol J Linn Soc. 56:483–503.
- Vaidya G, Lohman DJ, Meier R. 2011. SequenceMatrix: concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics. 27:171–180.
- Verma KK, Saxena R. 1996. The status of Bruchidae as a family. Chrysomela. 32:3.
- Verma KK. 1998. More on the bruchid controversy. Chrysomela. 35:3.
- Xie YL, Wu GX, Tang JB, Luo R, Patterson J, Liu S, Huang W, He G, Gu S, Li S, et al. 2014. SOAPdenovo-Trans: de novo transcriptome assembly with short RNA-Seq reads. Bioinformatics. 30:1660–1666.
- Zhou X, Li YY, Liu SL, Yang Q, Su X, Zhou LL, Tang M, Fu RB, Li JG, Huang QF. 2013. Ultra-deep sequencing enables high-fidelity recovery of biodiversity for bulk arthropod samples without PCR amplification. Gigascience. 2:4.