
Abstract
Camellia fangchengensis is endemic to Fangcheng, Guangxi Province, China, and its populations have been shrinking. In the present study, we report the complete chloroplast genome of C. fangchengensis using HiSeq 2500 sequencing technology. The complete chloroplast genome length is 157,095 bp. The chloroplast genome has 43 tRNA genes, six rRNA genes, and 97 protein-coding genes. Moreover, 18 genes have multiple copies in the chloroplast genome. A total of 146 genes are present in the chloroplast genome. Maximum likelihood phylogenetic tree based on 11 complete chloroplast genomes revealed that C. fangchengensis is closely related to C. pitardii. The complete chloroplast genome of C. fangchengensis would help to conserving the precious natural populations.
Camellia fangchengensis is found in Fangcheng, Guangxi Province, China. The occurrence of this species is estimated to be less than 100 km2, consisting of only one location. The last known herbarium specimen was collected in 1994, and several botanists have failed to relocate the species in the wild at the type locality. The red list category is Critically Endangered B1ab(iii) (http://www.iucnredlist.org/details/62057104/0). The genus Camellia possesses significant economic values (Yang et al. Citation2013; Huang et al. Citation2014), and it provides excellent materials for studying the interspecific hybridisation in plant sciences (Yang et al. Citation2013). Camellia fangchengensis might become a good material for studying functional genomics and provide a valuable breeding resource in the genus of Camellia. The chloroplast genome information which has been applied in plant biology, phylogenetic inference and species identification (Kane et al. Citation2012; Ruhfel et al. Citation2014). However, a total of 14 complete chloroplast genomes in the genus Camellia, excluding that of C. fangchengensis have been reported (Shi et al. Citation2013; Yang et al. Citation2013; Huang et al. Citation2014).
In the present study, we assembled the complete chloroplast genome sequence of C. fangchengensis using Illumina HiSeq2500 sequencing technology. We have deposited the complete chloroplast genome sequence into GenBank database (Accession Number: MG198672).
Young leaves of C. fangchengensis were collected from International Camellia Species Garden (29°07′21.60″ N, 119°35′34.98″ E). The specimens have been preserved in the laboratory of the Kunming Institute of Botany, The Chinese Academy of Sciences, and the accession number is CF201709. Total genomic DNA was extracted from young leaves (10 g) by CTAB method (Doyle and Doyle Citation1987).
Pair-end sequencing (Library insert size: 500 bp) was performed using the HiSeq 2500 platform following the manufacturer’s protocol (Illumina, San Diego, CA, USA). Trimmomatic v0.22 (Bolger et al. Citation2014) was used to quality control. Totally, 59.72 M plastid reads were filtered from sequencing reads with a sufficient coverage, plastid reads were assembled to preliminary chloroplast genome by SOAPdenovo2 (Luo et al. Citation2012). Twenty-nine gaps were filled by the polymerase chain reaction (PCR)-based methods and GapFiller (Nadalin et al. Citation2012). The chloroplast genome was annotated by the software DOGMA (Wyman et al. Citation2004). A physical map of the chloroplast genome was prepared using the OGDRAW web server (Lohse et al. Citation2013).
We performed a phylogenetic analysis using nine complete chloroplast genomes of Camellia species and Coffea arabica as an out-group. Eleven complete chloroplast genomes were aligned by clustalw-mpi version 0.13 (Li Citation2003). Maximum likelihood analysis was implemented with MEGA v7.0.26 (Kumar et al. Citation2016).
The chloroplast genome of C. fangchengensis was 157,095 bp, with a circular structure. The length of inverted repeats (IR), large single copy (LSC), and small single copy (SSC) was 13,420, 130,675, and 19,354 bp, respectively.
The chloroplast genome of C. fangchengensis contained six rRNA genes, 43 tRNA genes, and 97 protein-coding genes. Among them, 11 protein-coding genes (atpF, ndhA, ndhB, orf42, rpl2, rps12, rps7, ycf1, ycf15, ycf2, and ycf68), five tRNA (trnL-UAA, trnV-UAC, trnI-GAU, trnA-UGC, and trnI-GAU), and three rRNA genes (16SrRNA, 23SrRNA, and 5SrRNA) had multiple copies. Annotation revealed that six tRNA genes, seven protein-coding genes and three rRNA genes were duplicated in the IR region. Totally, 146 genes were present in the chloroplast genome of C. fangchengensis, and the G + C content was 37.19% (A = 31.17%, T = 31.64%, G = 18.21%, and C = 18.98%).
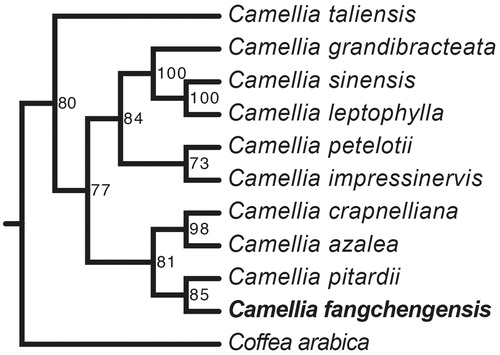
The maximum likelihood phylogenetic tree showed that C. fangchengensis was closely related to C. pitardii. Five species of Camellia formed a single cluster (C. grandibracteata, C. sinensis, C. leptophylla, C. petelotii, and C. impressinervis). Four species of Camellia formed another cluster (C. crapnelliana, C. azalea, C. pitardii, and C. fangchengensis). Camellia taliensis was located outside the clusters of Camellia ().
Figure 1. Maximum likelihood phylogenetic tree of Camellia fangchengensis based on 11 complete chloroplast genome sequences using Coffea arabica as an out-group. Numbers in the nodes are bootstrap values based on 1000 replicates. Accession numbers are listed as below: Camellia azalea NC_035574, Camellia crapnelliana NC_024541, Camellia grandibracteata NC_024659, Camellia impressinervis NC_022461, Camellia leptophylla NC_024660, Camellia petelotii NC_024661, Camellia pitardii NC_022462, Camellia sinensis NC_020019, Camellia taliensis NC_022264, Coffea arabica NC_008535.
The complete chloroplast genome of C. fangchengensis would provide information on phylogeny and species identity that would enhance the conservation strategies of Camellia in the future.
Disclosure statement
There are no conflicts of interest to declare.
Additional information
Funding
Related Research Data
References
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30:2114–2120.
- Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19:11–15.
- Huang H, Shi C, Liu Y, Mao SY, Gao LZ. 2014. Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: genome structure and phylogenetic relationships. BMC Evol Biol. 14:151–151.
- Kane N, Sveinsson S, Dempewolf H, Yang JY, Zhang D, Engels JM, Cronk Q. 2012. Ultra-barcoding in cacao (Theobroma spp.; Malvaceae) using whole chloroplast genomes and nuclear ribosomal DNA. Am J Bot. 99:320–329.
- Luo R, Liu B, Xie Y. 2012. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience. 1:18.
- Li K. 2003. ClustalW-MPI: ClustalW analysis using distributed and parallel computing. Bioinformatics. 19:1585–1586.
- Lohse M, Drechsel O, Kahlau S, Bock R. 2013. OrganellarGenomeDRAW-a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41:575–581.
- Nadalin F, Vezzi F, Policriti A. 2012. Gapfiller: a de novo assembly approach to fill the gap within paired reads. BMC Bioinformatics. 13 Suppl 14:): S8.
- Kumar S, Stecher G, Tamura K. 2016. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol. 33:1870–1874.
- Ruhfel BR, Gitzendanner MA, Soltis PS, Soltis DE, Burleigh JG. 2014. From algae to angiosperms – inferring the phylogeny of green plants (Viridiplantae) from 360 plastid genomes. BMC Evol Biol. 14:1–27.
- Shi C, Liu Y, Huang H, Xia EH, Zhang HB, Gao LZ. 2013. Contradiction between plastid gene transcription and function due to complex posttranscriptional splicing: an exemplary study of ycf15 function and evolution in angiosperms. Plos One. 8:e59620.
- Wyman SK, Jansen RK, Boore JL. 2004. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 20:3252–3255.
- Yang JB, Yang SX, Li HT, Yang J, Li DZ. 2013. Comparative chloroplast genomes of Camellia species. Plos One. 8:e73053.