
Abstract
The mitochondrial genomes from two individuals of the extinct subspecies of the Sevan trout Salmo ischchan danilewskii are published in this paper. The mitochondrial DNA (mtDNA) is 16,665 base pairs (bp) in length and contained 13 protein-coding genes, 2 rRNA genes, and 22 tRNA genes. The overall base composition of the genome in descending order was 27.9% of A, 29.4% of C, 16.7% of G, and 26.0% of T without a significant AT bias of 53.9%.
DNA samples from two individuals of the extinct subspecies Salmo ischchan danilewskii, were obtained from scales stored in an old scale book archived at the Sevan Hydrobiological Station (Sevan, Armenia). The scales were collected on 19 May 1975 in the Lake Sevan near Sarykaya (BO1 individual) (40.3603 N and 45.2379 E) and on 16 May 1974 in the Lake Sevan near vil. Tovak (BO6 individual) (40.1870 N and 45.6053 E) from fish caught by seine net.
DNA was extracted from old scales of Sevan trout in the aDNA facilities of the National Research Center ‘Kurchatov institute’ (Moscow, Russia), following the methodology described previously (Orlando et al. Citation2013). Two DNA libraries were prepared using an NEB Next Quick DNA Library Prep Master Mix set for 454 (New England Biolabs, Ipswich, MA) with adapter primers based on Illumina Sequencing Platform (Sarkissian et al. Citation2015). Mitochondrial genome was sequenced using Illumina Hiseq 1500 ((Illumina, San Diego, CA) with 150 bp paired-end reads.
205,942,786, and 108,390,940 Illumina paired-end reads were generated for DNA library of B01 sample and B06 sample, respectively. Illumina reads from two DNA libraries were mapped to the mitochondrial genome of S. trutta (JQ390057) using the bowtie2 software version 2.2.3 (Langmead and Salzberg Citation2012) with very-sensitive-local preset options. Sequences were aligned using multiple sequence alignment program Muscle 3.8.31 (Edgar Citation2004). All gaps and poorly aligned positions were removed using Gblocks 0.91b (Talavera and Castresana Citation2007), resulting in 16,665 bp length alignment.
As a result, the mitogenome of S. ischchan danilewskii consists of 16,665 bp (GenBank accession numbers B01: MG599465 and B06: MG599466) and includes 13 protein-coding genes (PCGs), 2 rRNA genes, and 22 tRNA genes.
Eleven of the 13 PCGs (NAD4, NAD5, NAD4L, NAD3, COB, NAD1, NAD2, COX2, ATP8, ATP6, and COX3) used ATG as start codon, another one (COX1) used GTG and NAD6 used ATA. Twelve genes (NAD1, NAD2, COX1, COX2, ATP8, ATP6, COX3, NAD3, NAD4L, NAD4, NAD5, and COB) ended with a TAA stop codon, but for three ones of them (COX2, NAD4, and COB) TAA stop codon is completed by the addition of 3′ A residues to the mRNA, NAD6 gene ended with a TAG stop codon.
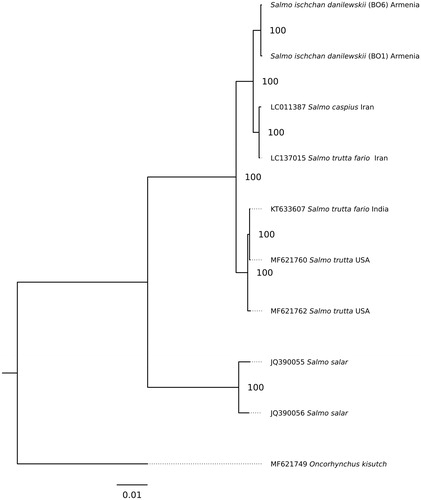
The phylogenetic analysis for whole mitogenome sequences was performed for S. ischchan danilewskii and other Salmonidae species: S. trutta fario (LC137015.1); S. trutta (MF621760.1); S. trutta (MF621762.1); S. salar (JQ390055.1); S. salar (JQ390056.1) and Oncorhynchus kisutch (MF621749.1) (). The phylogenetic relationships were reconstructed using the maximum-likelihood (ML) method in the PhyML 2.4.5 (Guindon and Gascuel Citation2003). The best substitution model (averaged for whole mitogenome) was chosen in the jModelTest 2.1.10 (Darriba et al. Citation2012) on the basis of the corrected Akaike information criterion (AICc). According to jModelTest, the best model describing the evolution of the mitogenomes was GTR + G (−lnL =34,558.09, AICc =69,170.28), and therefore, it was used for ML analysis ().
Figure 1. The Maximum-likelihood phylogenetic tree for S. ischchan danilewskii and other Salmonidae species.
Acknowledgements
The authors are grateful to Mikhail V. Kovalchuk (National Research Centre ‘Kurchatov Institute’, Moscow, Russia) for his ongoing support.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Additional information
Funding
Related Research Data
References
- Darriba D, Taboada GL, Doallo R, Posada D. 2012. jModelTest 2: more models, new heuristics and parallel computing. Nature Methods. 9:772
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research. 32:1792–1797.
- Guindon S, Gascuel O. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Systematic Biol. 52:696–704.
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nature Methods. 9:357–354.
- Orlando L, Ginolhac A, Zhang GJ, Froese D, Albrechtsen A, Stiller M, Schubert M, Cappellini E, Petersen B, Moltke I, et al. 2013. Recalibrating Equus evolution using the genome sequence of an early Middle Pleistocene horse. Nature. 499:74–78.
- Sarkissian CD, Ermini L, Schubert M, Yang MA, Librado P, Fumagalli M, Jonsson H, Bar-Gal GK, Albrechtsen A, Vieira FG, et al. 2015. Evolutionary genomics and conservation of the endangered Przewalski's horse. Current Biology. 25:2577–2583.
- Talavera G, Castresana J. 2007. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Systematic Biol. 56:564–577.