
Abstract
The complete mitochondrial genome of a marine fish Siganus sutor was completely sequenced by the high throughput sequencing method. This complete mitochondrial genome was 16,497 bp in length, consisted of 13 protein-coding genes, 22 tRNA genes, two rRNA genes and one large non-coding region. The gene arrangement of S. sutor is identical to those in typical fishes. Phylogenetic tree based on 13 protein-coding genes shows that Siganidae has a closer phylogenetic relationship to Luvaridae than to Ephippidae or Scatophagidae.
Siganus sutor, common name Shoemaker spinefoot, is a kind of coral reef fish (Grandcourt Citation2002). Long-distance movement of this fish have been reported (Kaunda-Arara and Rose Citation2004). They often occur among seagrasses to browse on ‘aufwuchs’ and form schools, and they occasionally eat poisonous fish (Gilbert Citation1993). The coloration of their body is highly variable; and influenced by substrate's colour and mood of the fish, upper colour is greenish to sandy brown, and the below is paler, with this colour patterns extending to the fins. In this study, we first reported the complete mitochondrial genome of S. sutor based on samples collected from Naozhou island in Zhanjiang, China (geographic coordinate: N 20°53′20.11″, E 112°28′46.2″), and analyzed its phylogenetic relationship with some other species from families Siganidae, Luvaridae, Scatophagidae, Ephippidae and Acanthuridae. The whole body specimen was preserved in ethanol and registered to the Marine Biodiversity Collection of South China Sea, Chinese Academy of Sciences, under the voucher number SCF20171022004.
The complete mitochondrial genome of S. sutor was 16,497 bp in length (GenBank accession No. MG677546), which included 13 protein-coding genes, two rRNA genes, 22 tRNA genes, one OL (origin of replication on the light-strand) and one D-Loop (control region). The OL was 50 bp in length, located in the cluster of five tRNA genes (WANCY region) between tRNA-Asn and tRNA-Cys. The D-loop was 831 bp in length, located between tRNA-Pro and tRNA-Phe. Gene arrangement of this genome was identical to that of those in typical fishes and most genes were encoded by the heavy strand (H-strand), except for ND6 and eight tRNA genes (Inoue et al. Citation2001; Wang et al. Citation2017). Overall base composition values for the mitochondrial genome were 28.04%, 29.82%, 16.75%, and 25.39% for A, C, G, and T, respectively.
The phylogenetic relationships of S. sutor with 12 closely related species were analyzed in this study. The complete mitochondrial genes of these 13 species were available on GenBank. The Maximum Likelihood evolutionary tree (ML tree) was constructed by MEGA 7 (Kumar et al. Citation2016) based on 1st and 2nd codon sequences of 13 protein-coding genes.
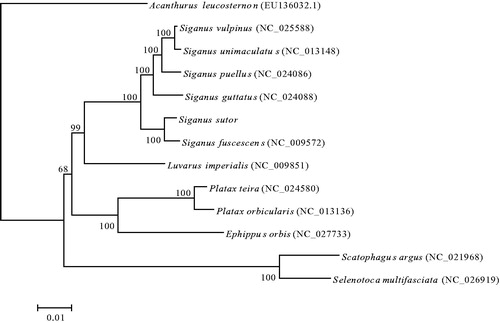
In the ML phylogenetic tree, compared with other species, S. sutor and S. fuscescens have the closest phylogenetic relationship, and clade with S. guttatus, S. puellus, S. unimaculatus and S. vulpinus with a strong support. And these five species were all classified into family Siganidae of order Perciformes. Luvarus imperialis is a sister lineage to the formerly mentioned clade, Ephippus orbis, Platax orbicularis and Platax teira formed another clade, which was classified into family Ephippidae of order Perciformes. Another clade included Scatophagus argus and Selenotoca multifasciata with a strong support, which was classified into family Scatophagidae of order Perciformes (). These results show that phylogenetic relationship between Siganidae and Luvaridae was closer than to Ephippidae and Scatophagidae.
Figure 1. Maximum likelihood phylogenetic tree was constructed based on 1st and 2nd codon sequences of 13 protein-coding genes of 13 species.
Disclosure statement
The authors declare that there is no conflict of interest regarding the publication of this article. The authors alone are responsible for the content and writing of the paper.
Additional information
Funding
Related Research Data
References
- Gilbert CR. 1993. World fishes important to North Americans, exclusive of species from the continental waters of the United States and Canada. Rev Fish Biol Fisheries. 3:191–192.
- Grandcourt EM. 2002. Demographic characteristics of a selection of exploited reef fish from the Seychelles: preliminary study. Mar Freshwater Res. 53:123–130.
- Inoue JG, Miya M, Aoyama J, Ishikawa S, Tsukamoto K, Nishida M. 2001. Complete mitochondrial DNA sequence of the Japanese eel Anguilla japonica. Fisheries Sci. 67:118–125.
- Kaunda-Arara B, Rose GA. 2004. Long-distance movements of coral reef fishes. Coral Reefs. 23:410–412.
- Kumar S, Stecher G, Tamura K. 2016. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol Biol Evol. 33:1870–1874.
- Wang PF, Chen SX, Ou YJ, Wen JF, Li JE. 2017. Complete mitochondria! genome of Toxabramis houdemeri (Cypriniformes; Cyprinidae) and its phylogenetic analysis. Mitochondrial DNA A. 28:292–293.