
Abstract
The bamboo snout beetle Cyrotrachelus buqueti (Coleoptera: Curculionidae) is a destructive forest pest and distributed widely in Southeast Asia. The 15,035 bp complete mitochondrial genome of the species consists of 13 protein-coding genes (PCGs), two ribosomal RNA genes (rRNAs), 21 transfer RNA genes (tRNAs) and a control region (GenBank accession no. MG674390). The trnl gene was not found in the C. buqueti mitogenome. The gene order and the orientation of C. buqueti were similar to those found in other Coleoptera species. The nucleotide composition was significantly biased (A, G, C, and T was 38.18%, 10.10%, 16.16%, and 35.56%, respectively) with A + T contents of 73.74%. ATG, ATA, ATT, AAT, and TTG were initiation codons and TAA, TAG, and T were termination codons. All the 21 tRNAs displayed a typical cloverleaf secondary structure, except for trnS1 which lacked the dihydrouridine arm. Phylogenetic analysis was performed using 13 PCGs with 14 other beetles showed that C. buqueti is closely related to Eucryptorhynchus brandti, which agree with the traditional classification.
The bamboo snout beetle, Cyrtotrachelus buqueti Guerin-Meneville (Coleoptera: Curculionidae), is a highly destructive forest pest and distributed widely in Southeast Asia (Yang et al. Citation2017). The larvae of C. buqueti bore into the shoots of clumping bamboo species, causing serious damage to bamboo production (Yang et al. Citation2009). Adult specimens of C. buqueti were collected from Nanming District, Guiyang City, Guizhou Province, China (N26°33′, E106°46′). Samples have been deposited in the insect specimen room of Guiyang University with an accession number GYU-Col-20170001).
The circular mitochondrial genome of C. buqueti was 15,035 bp in length (GenBank accession no. MG674390) and included sets of genes, including 13 protein-coding genes (PCGs), two ribosomal RNA genes (rrnL and rrnS), 21 transfer RNA genes (tRNAs), and a large non-coding region (putative control region). The trnl was not found in the C. buqueti mitogenome, as observed in Sympiezomias velatus (Tang et al. Citation2017), another completely sequenced species in Coleoptera. The gene order and the orientation of C. buqueti were similar to those of putative ancestor of insects (Boore Citation1999). The nucleotide composition of was significantly biased (A, G, C, and T was 38.18%, 10.10%, 16.16%, and 35.56%, respectively) with A + T contents of 73.74%. The AT-skew and GC-skew of this genome were 0.036 and −0.231, respectively. Twenty-two genes were transcribed on the J-strand, whereas the others were oriented on the N-strand. Gene overlaps were present at eight gene junctions and involved a total of 20 bp; the longest overlap (7 bp) existed between atp8 and atp6. A total of 59 bp intergenic spacers were found in 10 positions, ranging in size from 1 to 21 bp. The largest intergenic spacer sequence of 21 bp was located between trnS2 and nad1. The control region was located between rrnS and trnQ gene with a length of 430 bp, and the A + T content was 81.16%.
All the 21 tRNAs were predicted to have typical cloverleaf secondary structures, except the gene trnS1 (AGN) lacking a stable dihydrouridine arm, which were consistent with those reported in other animal mitogenomes (Wolstenholme Citation1992; Yuan et al. Citation2016). The length of these tRNAs ranged from 63 bp (trnC and trnE) to 71 bp (trnK), A + T content ranged from 58.46% (trnN) to 86.15% (trnD). The rrnL was located between trnL1 and trnV, and rrnS resided between trnV and the control region. The rrnL was 1298 bp in length with A + T content of 78.43%, and the rrnS was 788 bp in length with A + T content of 75.13%.
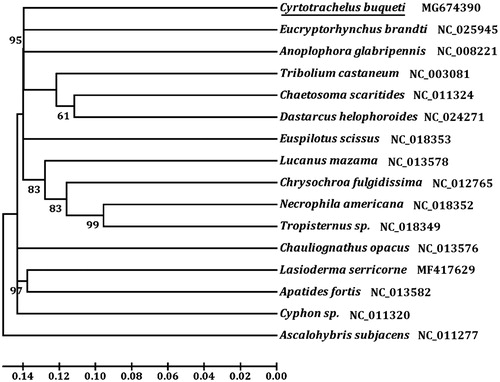
The initial codons for 11 PCGs of C. buqueti were the canonical putative start codons ATN (ATG for atp6, nad4L, and cob; ATT for cox2, atp8, and nad5; ATA for cox3, nad3, nad1, and nad4; ATC for nad2). However, cox1 and nad6 used AAT and TTG as start codon, respectively. Nine PCGs were terminated with TAA or TAG, and the remaining PCGs including cox1, cox3, nad4, and nad5 use a single T as stop codon. We analyzed the amino acid sequences of 13 PCGs with neighbour-joining method to construct the phylogenetic relationship of C. buqueti with 14 other representative bettles. The result showed that C. buqueti is closely related to Eucryptorhynchus brandti (), which agree with the traditional classification.
Figure 1. Phylogenetic tree showing the relationship between C. buqueti and 14 other beetles based on neighbour-joining method. Ascalohybris subjacens was used as an outgroup. GeneBank accession numbers of each species were listed in the tree.
Disclosure statement
The authors report no conflicts of interests. The authors alone are responsible for the content and writing of the paper.
Additional information
Funding
References
- Boore JL. 1999. Survey and summary: animal mitochondrial genomes. Nucleic Acids Res. 27:1767–1780.
- Wolstenholme DR. 1992. Animal mitochondrial DNA: structure and evolution. Int Rev Cytol. 141:173–216.
- Tang PA, Zhang L, Li XP, Li FF, Yuan ML. 2017. The complete mitogenome of Sympiezomias velatus (Coleoptera: Curculionidae). Mitochondrial DNA Part B. 2:449–450.
- Yang H, Su T, Yang W, Yang C, Lu L, Chen Z. 2017. The developmental transcriptome of the bamboo snout beetle Cyrtotrachelus buqueti and insights into candidate pheromone-binding proteins. PLoS ONE. 12:e0179807.
- Yang YJ, Wang SF, Gong JW, Liu C, Mu C, Qin H. 2009. Relationships among Cytotrachelus buqueti larval density and wormhole number and bamboo shoot damage degree. Chinese J Appl Ecol. 20:1980–1985.
- Yuan ML, Zhang QL, Zhang L, Guo ZL, Liu YJ, Shen YY, Shao RF. 2016. High-level phylogeny of the Coleoptera inferred with mitochondrial genome sequences. Mol Phylogenet Evol. 104:99–111.