
Abstract
Leucanthemella linearis is an important marsh plant. The complete chloroplast genome sequence of L. linearis was obtained using next generation sequencing. It was 15,1401 bp in length, including a pair of inverted repeat (IR, 24,941 bp) regions separated by a small single copy (SSC, 18,392 bp) sequence and a large single copy (LSC, 83,127 bp) sequence. The cp genome contained 140 genes, consisting of 96 protein-coding genes, 8 rRNA genes and 36 tRNA genes. Twenty-two genes were present in the IR region. Thirty-four SSR sites were detected in the cp genome. Phylogenetic analysis with the reported chloroplast genomes revealed that L. linearis is most closely related to Tribe Heliantheae species. This new data will help to understand the phylogenetic position and biology of the Leucanthemella.
The growth of the population and the acceleration of industrialization destroyed the ecological environment. The wetland areas have shrunk gradually and the biodiversity of wetland is decreasing and some species are even extinct which thus threatened our sustainability (Niu et al. Citation2012; Zhou et al. Citation2014).
Leucanthemella linearis (Matsum.) Tzvel. (2n = 18) is distributed in East Asia from Manchuria, China, Korea, to Japan and grows in marsh areas. As perennial plants, L. linearis has the pretty scene value for these wetlands. Here we obtained the complete chloroplast genome of L. linearis using the Illumina HiSeq platform. The cp genome sequence of L. linearis will not only protect the genetic resource of wetland, but also help to clarify phylogenetic relationships of the Tribe Anthemideae.
Fresh leaves of L. linearis were collected from Hani wetland (Jilin, China) and the voucher specimen was deposited into Herbarium of Northwest A&F University (China). Total genomic DNA was extracted from fresh leaves using CTAB method (Doyle and Doyle Citation1987) with some modification.
Then the DNA was sent to GENEWIZInc (Jiangsu, China) for library construction and sequencing. Paired-end reads of 2 × 300 were generated on an Illumina MiSeq Sequencer. Approximately 3.0 Gb of clean reads data were generated for L. linearis after trimming with Trimmomatic v0.36 (http://www.sadellab.org/cms/index.php?page=trimmomatic) (Bolger et al. Citation2014). De novo and reference-guided methods were combined to assemble chloroplast genome using Geneious10.1.3 (Kearse et al. Citation2012). The complete chloroplast genomes were annotated using the webserver DOGMA (Wyman et al. Citation2004) and the sequence was deposited in GenBank with the accession number MG748695. Simple sequence repeat (SSR) was investigated using Gramene (http://www.gramene.org/db/markers/ssrtool) (Temnykh et al. Citation2001). The complete cp genome of L. linearis was 151,401 bp in length, including a pair of inverted repeat (IR, 24,941 bp) regions seperated by a small single copy (SSC, 18,392 bp) sequence and a large single copy (LSC, 83,127bp) sequence (). The GC content of cp genome in the L. linearis is 37.3% and the corresponding values in LSC, SSC and IR regions are 35.4, 30.7 and 43%, respectively. The complete cp genome contained 140 genes, including of 96 protein-coding genes, 8 rRNA genes and 36 tRNA genes. Twenty-two genes were present in the IR region. Thirty-four SSR sites were detected in the cp genome of L. linearis. Most of them were composed of A or T base and dimer and tetramer were the majority.
Figure 1. Bayesian 50% majority-rule consensus tree of L. linearis with 5 Asteraceae species inferred from whole chloroplast genomes. Bayesian posterior probabilities are shown above the branches.
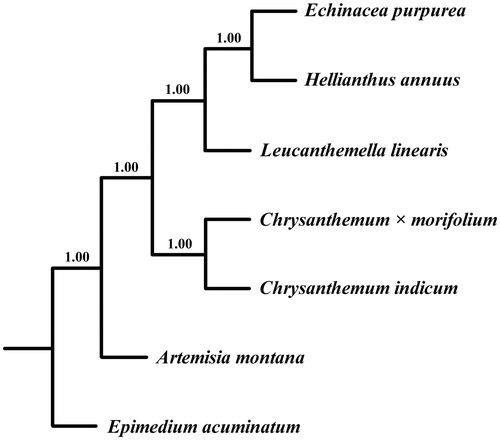
The phylogenetic relationship between L. linearis and related species was performed with MrBayes3.2.1 (Ronquist et al. Citation2012) using six chloroplast genomes of Asteraceae aligned with MAFFT (Katoh and Toh Citation2010). Epimedium acuminatum was added as the outgroup. The phylogenetic analysis reveals that L. linearis was closely related to Helianthus tribe species (). The chloroplast resource will provide molecular genetic information for DNA barcoding, conservation genetics, and breeding of L. linearis in the future.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30:2114–2120.
- Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19:11–15.
- Katoh K, Toh H. 2010. Parallelization of the MAFFT multiple sequence alignment program. Bioinformatics. 26:1899–1900.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28: 1647–1649.
- Niu ZG, Zhang HY, Wang XW, Yao WB, Zhou DM, Zhao KY, Zhao H, Li NN, Huang HB, Li CC, et al. 2012. Mapping wetland changes in China between 1978 and 2008. Chinese Sci Bull. 57:1400–1411.
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Hohna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61:539–542.
- Temnykh S, DeClerck G, Lukashova A, Lipovich L, Cartinhour S, McCouch S. 2001. Computational and experimental analysis of microsatellites in rice (Oryza sativa L.): frequency, length variation, transposon associations, and genetic marker potential Source. Genome Res. 11: 1441–1452.
- Wyman SK, Jansen RK, BooreJ L. 2004. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 20:3252–3255.
- Zhou SL, Zou XH, Zhou ZQ, Liu J, Xu C, Yu J, Wang Q, Zhang DM, Wang XQ, Ge S, et al. 2014. Multiple species of wild tree peonies gave rise to the ‘king of flowers’, Paeonia suffruticosa Andrews. Proc Biol Sci. 28:pii: 20141687.