
Abstract
The two complete mitochondrial genomes of endangered form of the Sevan trout Salmo ischchan aestivalis are published in this paper. The mitochondrial DNA (mtDNA) is 16,677 base pairs (bp) in length and contained 13 protein-coding genes, 2 rRNA genes, and 22 tRNA genes. The overall base composition of the genome in descending order was 29.4% – C, 27.9% – A, 26.0% – T, 16.7% – G, without a significant AT bias of 53.9%.
Main text
DNA samples of the Sevan trout, Endangered subspecies Salmo ischchan aestivalis, were obtained from two individuals: Li4 individual was collected from local aquaculture farm near Djermuk town on 2012 (39.7305 N and 45.5950 E) and Li30 was collected from the natural population, the Makenis River, tributary of Lake Sevan, on 6 May 1974 (40.1771 N and 45.5841 E). Li4 and Li30 samples were deposited at Ichthyological collection of Papanin Institute for Biology of Inland Waters, Russian Academy of Sciences, Borok, Yaroslavl, Russia. For the latter specimen, we used historical scales from scale book, a DNA source that is appropriate for mitogenome sequencing by NGS technology (Nedoluzhko et al. Citation2018).
DNA was isolated from caudal fin (Li4) and dried scales (Li30) in the DNA facilities of the National Research Center ‘Kurchatov institute’ (Moscow, Russia) using a standard method of DNA extraction from animal tissue (phenol-chloroform). The concentration of DNA in the samples was measured using a Qubit fluorimeter (Thermo Fisher Scientific, Waltham, MA, USA).
Two DNA libraries were prepared using an Ovation Ultralow Systems V2 kit (NuGEN, San Carlos, CA, USA). Mitochondrial genome was sequenced used Illumina Hiseq 2500 (Illumina, San Diego, CA, USA) with 150 bp pared-end reads.
33,981,829 and 144,017,525 Illumina paired-end reads were generated for DNA library of Li4 sample and Li30 sample, respectively. Illumina reads from Li30 sample were used for building mtDNA sequence de-novo by Norgal software package (Al-Nakeeb et al. Citation2017), but for Li4 library, there were not enough reads for building whole mitochondrial genome continuous contig. The Li4 reads were mapped to the Li30 mitochondrial genome using the Bowtie2 software version 2.2.3 with – very-sensitive-local preset options (Langmead and Salzberg Citation2012).
As a result, the mitogenome of S. ischchan aestivalis consists of 16,677 bp (GenBank accession numbers Li4: MG599465 and Li30: MG599466) and includes 13 protein coding genes (PCGs), 2 rRNA genes and 22 tRNA genes.
Eleven of the 13 PCGs (NAD4, NAD5, NAD4L, NAD3, COB, NAD1, NAD2, COX2, ATP8, ATP6, COX3) used ATG as start codon, another one (COX1) used GTG and NAD6 used ATA. Twelve genes (NAD1, NAD2, COX1, COX2, ATP8, ATP6, COX3, NAD3, NAD4L, NAD4, NAD5, and COB) ended with a TAA stop codon, but for three of them (COX2, NAD4, and COB), TAA stop codon is completed by the addition of 3′ A residues to the mRNA, NAD6 gene ended with a TAG stop codon.
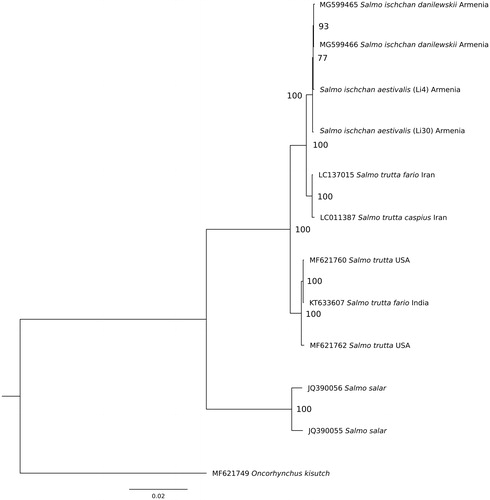
The phylogenetic analysis for whole mitogenome sequences was performed for the S. ischchan aestivalis and other Salmonidae species: S. ischchan danilewskii (MG599465 and MG599466), S. trutta fario (LC137015); S. trutta (MF621760); S. trutta (MF621762); S. salar (JQ390055); S. salar (JQ390056), and Oncorhynchus kisutch (MF621749) (). Sequences were aligned using multiple sequence alignment program Muscle 3.8.31 (Edgar Citation2004). All gaps and poorly aligned positions were removed using Gblocks 0.91b (Talavera and Castresana Citation2007), resulting in 16,511 bp length alignment. The phylogenetic relationships were reconstructed using the maximum likelihood (ML) method in the PhyML 2.4.5 (Guindon and Gascuel Citation2003). The best substitution model (averaged for whole mitogenome) was chosen in the jModelTest 2.1.10 (Darriba et al. Citation2012) on the basis of the Bayesian information criterion (BIC). According to jModelTest, the best model describing the evolution of the mitogenomes was TVM + G (–lnL = 34659.93, BIC = 69611.21), and therefore, it was used for ML analysis ().
Figure 1. The maximum-likelihood phylogenetic tree for Salmo ischchan aestivalis and other salmonid species.
Acknowledgments
The authors are grateful to Mikhail V. Kovalchuk (National Research Centre ‘Kurchatov Institute’, Moscow, Russia) for his ongoing support.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Al-Nakeeb K, Petersen TN, Sicheritz PT. 2017. Norgal: extraction and de novo assembly of mitochondrial DNA from whole-genome sequencing data. BMC Bioinformatics. 18:510.
- Darriba D, Taboada GL, Doallo R, Posada D. 2012. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 9:772–772.
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797.
- Guindon S, Gascuel O. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Systematic Biol. 52:696–704.
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods. 9:357–U354.
- Nedoluzhko AV, Rastorguev SM, Simonov E, Boulygina ES, Sharko FS, Tsygankova SV, Gabrielyan BK, Roubenyan HR, Levin BA. 2018. Two complete mitochondrial genomes of extinct form of the Sevan trout Salmo ischchan danilewskii. Mitochondrial DNA B. 3:40–41.
- Talavera G, Castresana J. 2007. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Systematic Biol. 56:564–577.