
Abstract
Carya kweichowensis is an importantly economic tree species in Juglandaceae, which is critically endangered and endemic to Guizhou, China. The plastid genome of C. kweichowensis was assembled and characterized based on Illumina pair-end sequencing data using genome skimming approach. The complete plastid genome is 175,313 bp in length, with a GC content of 35.8%. It is a typical quadripartite structure, containing a large single copy (LSC) region of 89,858 bp and a small single copy (SSC) region of 3569 bp separated by a pair of extremely extensive inverted repeats (IRs) of 40,943 bp. A total of 142 genes were annotated, including 94 protein-coding genes, 40 tRNA genes, and eight rRNA genes. A maximum likelihood (ML) phylogenetic tree on plastid genomes revealed that C. kweichowensis is close to Annamocarya sinensis. The newly characterized complete plastid genome of C. kweichowensis will provide essential data for further studies of this endangered species.
The Guizhou hickory, Carya kweichowensis Kuang & A. M. Lu ex Chang & Lu, is a deciduous tree species with pinnately compound leaves and small nuts, narrowly scattered in subtropical evergreen broad-leaf forest and endemic to Guizhou, China (Zhang et al. Citation2013). However, due to its highly economic values of high-quality timber and nutritious nuts, the wild resources of C. kweichowensis have been anthropogenically over-exploited, and its natural habitats were fragmented. Recently, C. kweichowensis was ranked as critically endangered (CR) of the Red List of China Higher Plants based on IUCN Red List Categories and Criteria (Qin et al. Citation2017). The native populations of the species have dramatically declined in past decades, which need extremely urgent conservation. Hence it is important to set up appropriate conservation strategies to conserve the genetic diversity of the species.
In this study, we assembled and characterized the complete plastid genome of C. kweichowensis via genome skimming approach (Straub et al. Citation2012). Sequence data have been deposited in GenBank (Accession Number: MH121170).
We sampled a natural population with fresh, healthy leaves of C. kweichowensis from Anlong county of Guizhou, China (N 25°02′19″, E 105°14′44″). A single individual was selected for total genomic DNA isolation using CTAB method (Doyle and Doyle Citation1987). Voucher specimen (Liuj167640) was deposited in the Herbarium of Kunming Institute of Botany (KUN), Chinese Academy of Sciences. Illumina paired-end (PE) library was constructed from fragmented genomic DNA following the standard protocols (NEBNext® Ultra II™DNA Library Prep Kit for Illumina®), and sequenced on the Illumina HiSeq X Ten platform (Illumina, San Diego, CA). Approximately, 2.2 Gb of high quality clean data with adaptors trimmed and quality filtered were generated. The plastid genome PE reads were aligned to the reference (GenBank Accession: KX703001) using bowtie2 v2.2.6 (Langmead and Salzberg Citation2012), and assembled into a genome using SPAdes v3.9.0 (Bankevich et al. Citation2012). The annotation of the assembled plastid genome was performed with Dual Organellar Genome Annotator (DOGMA) (Wyman et al. Citation2004).
The complete plastid genome of C. kweichowensis was 175,313 bp in length, showing a typical quadripartite structure: a pair of inverted repeats (IRs) of 40,943 bp separating by a large single copy (LSC) region of 89,858 bp and a small single copy (SSC) region of 3569 bp. The GC content of the plastid genome was 35.8%. This plastid genome contained 142 genes, including 94 protein-coding genes, eight ribosomal RNA (rRNA) genes, and 40 transfer RNA (tRNA) genes. The phylogenetic relationship of C. kweichowensis was conducted based on the complete plastid genome sequences of 19 species in Fagales (Table S1), and the maximum-likelihood (ML) analysis was conducted using RAxML v 8.0 (Stamatakis Citation2014). The phylogenetic relationships of the 20 species were well resolved with strongly supported ().
Figure 1. Maximum-likelihood (ML) phylogenetic tree based on complete plastid genome sequences of the 20 species.
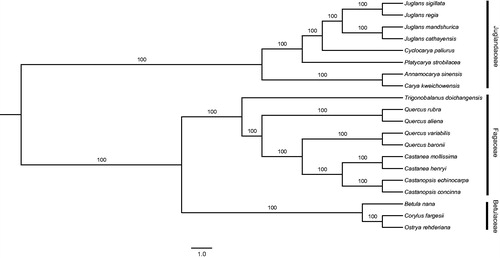
The newly characterized complete plastid genome of C. kweichowensis will provide essential resources for further study on the evolution and genetic diversity of the endemic species, as well as provide fundamental information to effectively conserve the CR species.
Acknowledgements
Laboratory work was carried out at the Laboratory of Molecular Biology in the Germplasm Bank of Wild Species in Southwest China, Kunming Institute of Botany, Chinese Academy of Sciences.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Pyshkin AV. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19:455–477.
- Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull Bot Soc Am. 19:11–15.
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods. 9:357–359.
- Qin HN, Yang Y, Dong SY, He Q, Jia Y, Zhao LN, Yu SX, Liu HY, Liu B, Yan YH, et al. 2017. Threatened species list of China’s higher plants. Biodivers Sci. 25:696–744.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30:1312–1313.
- Straub SC, Parks M, Weitemier K, Fishbein M, Cronn RC, Liston A. 2012. Navigating the tip of the genomic iceberg: next-generation sequencing for plant systematics. Am J Bot. 99:349–364.
- Wyman SK, Jansen RK, Boore JL. 2004. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 20:3252–3255.
- Zhang JB, Li RQ, Xiang XG, Manchester SR, Lin L, Wang W, Wen J, Chen ZD. 2013. Integrated fossil and molecular data reveal the biogeographic diversification of the eastern Asian-eastern North American disjunct hickory genus (Carya Nutt.). PLoS ONE. 8:e70449.