
Abstract
The complete chloroplast genome sequence of Caltha palustris, a species of the Ranunculaceae family, was characterized from the de novo assembly of HiSeq (Illumina Co.) paired-end sequencing data. The chloroplast genome of C. palustris was 155,292 bp in length, with a large single-copy (LSC) region of 84,120 bp, a small single-copy (SSC) region of 18,342 bp, and a pair of identical inverted repeat regions (IRs) of 26,415 bp. The genome contained a total of 114 genes, including 80 protein-coding genes, 30 transfer RNA (tRNA) genes, and 4 ribosomal RNA (rRNA) genes. The phylogenetic analysis of C. palustris with 14 related species revealed the closest taxonomical relationship with Hydrastis canadensis in the Ranunculaceae family.
The genus Caltha consists of approximately 16 perennial flowering plants belonging to the Ranunculaceae family, which habitats throughout the Northern and Southern Hemispheres (Schuettpelz and Hoot Citation2004). C. palustris, also known as marsh-marigold, has a poisonous characteristic in the leaves (Darbyshire et al. Citation2000). Morphologically, C. palustris has been indistinguishable from Ligularia fischeri, a widely used esculent herb in Korea. Therefore, the acquisition of chloroplast DNA information from C. palustris is important for future DNA barcode marker development to be distinguished from edible L. fischeri.
The leaves of C. palustris were provided from Hantaek botanical garden (www.hantaek.co.kr) in Yongin-si, Korea (37° 05′ 40.4″ N, 127° 24′ 23.7″ E) and used to construct the genomic library for Illumina paired-end (PE) sequencing. The high-quality PE reads were assembled by CLC Genomics Workbench (ver. 10.0.1, CLC QIAGEN), followed by manual curation through PE reads mapping (Kim et al. Citation2015). Annotation of the complete chloroplast genome was performed with GeSeq and manual corrections (Tillich et al. Citation2017). The complete chloroplast genome sequence of C. palustris was submitted to GenBank with the accession number of MG581742.
The complete chloroplast genome of C. palustris was 155,292 bp in length with 38.14% of G + C content, comprising a large single copy (LSC) region of 84,120 bp, a small single copy (SSC) region of 18,342 bp, and a pair of inverted repeat (IRa and IRb) regions of 26,415 bp. The genome contained 114 genes including 80 protein-coding genes, 30 tRNA genes, and 4 rRNA genes. In addition, 49 simple sequence repeats (SSR) were detected with the minimum repeat number of 10, 6, 5, 5, 5, and 5 for mono-, di-, tri-, tetra-, penta-, and hexa-nucleotides, respectively, using SSR-identification program (Beier et al. Citation2017).
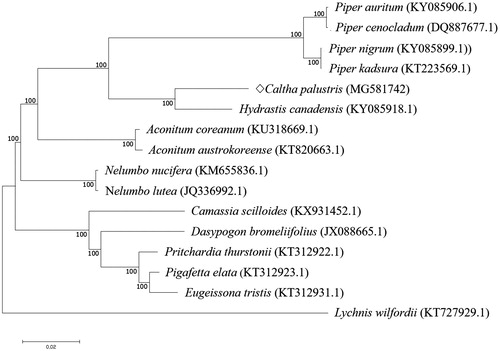
In order to investigate the evolutionary relationship, the complete chloroplast genome sequences of C. palustris and 14 related species were aligned using MAFFT (ver. 7.271) (Katoh et al. Citation2002), followed by phylogenetic tree construction obtained from a Maximum Likelihood (ML) analysis with 1000 bootstraps using MEGA 7.0 (Kumar et al. Citation2016). The phylogenetic tree exhibited the close relationship of C. palustris with Hydrastis canadensis in the family of Ranunculaceae ().
Figure 1. Phylogenetic analysis of C. palustris with 14 related species by Neighbor-Joining (NJ) methods. Phylogenetic tree was generated using complete chloroplast genome sequences, including outgroup species of Lychnis wilfordii. Numbers in the nodes are the bootstrap values from 1000 replicates.
Acknowledgements
We appreciate Hantaek Botanical Garden, Korea, for providing us plant material of Caltha palustris.
Disclosure statement
The authors report no conflict of interest.
This research was supported by a grant [17162MFDS065] from Ministry of Food and Drug Safety, Republic of Korea in 2018.
References
- Beier S, Thiel T, Munch T, Scholz U, Mascher M. 2017. MISA-web: a web server for microsatellite prediction. Bioinformatics. 33:2583–2585.
- Darbyshire SJ, Favreau M, Murray M. 2000. Common and Scientific names of weeds in Canada. Ottawa (ON): Agriculturue and Agri-Food Canada.
- Katoh K, Misawa K, Kuma K, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30:3059–3066.
- Kim K, Lee SC, Lee J, Yu Y, Yang K, Choi BS, Koh HJ, Waminal NE, Choi HI, Kim NH, et al. 2015. Complete chloroplast and ribosomal sequences for 30 accessions elucidate evolution of Oryza AA genome species. Sci Rep. 5:15655
- Kumar S, Stecher G, Tamura K. 2016. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 33:1870–1874.
- Schuettpelz E, Hoot SB. 2004. Phylogeny and biogeography of Caltha (Ranunculaceae) based on chloroplast and nuclear DNA sequences. Am J Bot. 91:247–253.
- Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, Greiner S. 2017. GeSeq – versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45:W6–W11.