
Abstract
Linnaea chinensis (Caprifoliaceae), which inhabits in China and Japan, is commonly cultivated as an ornamental shrub. The complete chloroplast genome of L. chinensis was newly assembled in this study based on Illumina pair-end sequencing data. The full length of L. chinensis plastome is 155,813 bp. In total, 124 genes were identified, including 75 protein-coding genes, 8 rRNA genes, and 42 tRNA genes. The overall GC content of this genome was 38.4%. A further phylogenomic analysis including16 species from Adoxaceae and Caprifoliaceae was constructed.
Linnaea chinensis A.Braun & Vatke is a compact deciduous shrub with funnel-shaped, white flowers. It is distributed in China and Japan, and is a horticulturally important species in eastern Asia. Linnaea chinensis is one of the parental species of the garden hybrid Linnaea × grandiflora. This species was recently transferred from genus Abelia based on molecular evidence (Christenhusz Citation2013). No chloroplast genome information was reported so far for this genus. In this article, we assembled and characterized the complete chloroplast genome of L. chinensis. It will provide essential genetic data for molecular breeding and phylogenomic analysis in this genus.
The sample was collected from Chun’an, Zhejiang, China (Voucher No. ZSTU00877, deposited at Zhejiang Sci-Tech University). Total DNA was extracted from fresh leaves of L. chinensis individual using DNA Plantzol Reagent (Invitrogen, Carlsbad, CA, USA). The plastome sequences were generated using Illumina HiSeq 2500 platform (Illumina Inc., San Diego, CA, USA). In total, ca. 30.8 million high-quality clean reads (150 bp PE read length) were generated with adaptors trimmed. Then, CLC Genomics Workbench (CLC Bio, Aarhus, Denmark) was used for trimming, de novo assembly, and mapping to Kolkwitzia amabilis (KT966716) as reference. BLAST, GeSeq (Tillich et al. Citation2017), tRNAscan-SE v1.3.1 (Schattner et al. Citation2005), and GENEIOUS v 11.0.5 (Biomatters Ltd, Auckland, New Zealand) were used to align, assemble, and annotate the plastome.
The full length of L. chinensis chloroplast genome (GenBank Accession No. MH553932) was 155,813 bp. It comprises large single-copy region (LSC with 89,954 bp), a small single-copy region (SSC with 18,761 bp), and two inverted repeat regions (IR with 46,906 bp). The overall GC content of the L. chinensis cp genome was 38.4%. A total of 124 genes were contained in the cp genome (75 protein-coding genes, 8 rRNA genes, and 42 tRNA genes). Two genes of tRNA (trnH-GUG, trnG-UCC) had four copies and one of tRNA (trnR-ACG) has three copies. Ten genes had two copies, which included 1 PCG genes (ndhB), 5 tRNA genes (trnL-CAA, trnV-GAC, trnI-GAU, trnA-UGC, trnN-GUU), and all 4 rRNA species (rrn4.5, rrn5, rrn16, and rrn23). Among the protein-coding genes, two genes (rps18 and ycf3) contained two introns, and other six genes (atpF, ndhA, ndhB, rpl2, rpoC1, rps16) had one intron each.
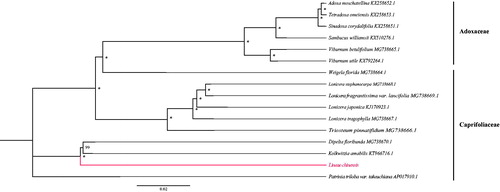
Using MAFFT v7.3 (Katoh and Standley Citation2013), we aligned 16 chloroplast genomes of species from Adoxaceae and Caprifoliaceae. A phylogenetic tree was drawn by statistical method of the maximum likelihood (ML) inference using GTR model with 1000 bootstrap replicates with RAxML v.8.2.1 (Stamatakis Citation2014) on the CIPRES cluster service (Miller et al. Citation2010). The result revealed that L. chinensis is sister to Dipelta floribunda and Kolkwitzia amabilis in Caprifoliaceae (). We hope the newly characterized complete chloroplast genome of L. chinensis will provide essential genetic resource and background data for further phylogenetic study and breeding practices of the genus Linnaea.
Figure 1. RAxML phylogeny of Linnaea based on 16 complete cp genomes (accession numbers were listed behind their names, and labels showed beside branches represent of bootstrap values, and “*” indicates a 100% bootstrap value).
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper
Additional information
Funding
References
- Christenhusz MJM. 2013. Twins are not alone: a recircumscription of Linnaea (Caprifoliaceae). Phytotaxa. 125:25–32.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30:772–780.
- Miller MA, Pfeiffer W, Schwartz T. 2010. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. Proceedings of the gateway computing environments workshop (GCE); New Orleans, LA. p. 1–8.
- Schattner P, Brooks AN, Lowe TM. 2005. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33:W686–W689.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30:1312–1313.
- Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, Greiner S. 2017. GeSeq – versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45:W6–W11.