
Abstract
In this study, the complete mitochondrial genome of Eastern diamondback rattlesnake, Crotalus adamanteus, was recovered through Illumina sequencing data. This complete mitochondrial genome of C. adamanteus is 17,406 bp in length and has a base composition of A (32.3%), T (24.7%), C (29.7%), G (13.3%), demonstrating a bias of higher AT content (57.0%) than GC content (43.0%). The mitochondrial genome contains a typically conserved structure among Crotalinae mitogenomes, encoding 13 protein-coding genes (PCGs), 22 transfer RNA genes (tRNA), 2 ribosomal RNA genes (12S rRNA and 16S rRNA) and a control region (D-loop region). Twelve PCGs were located on the H-strand, except ND6 that was located on the L-strand. ATP8 gene and ATP6 gene were overlapped by 9 bp. The whole mt genome of C. adamanteus and other Viperidae mitogenomes (44 species, in total) were used for phylogenetic analysis. The result indicated C. adamanteus clustered with Crotalus horridus (HM641837.1), representing a distinct genus, which has the closest relationship with Bothrops jararaca (KU194299.1).
The eastern diamondback rattlesnake (Crotalus adamanteus) is one of the largest pit viper native to the southeastern United States and is the largest member of the genus Crotalus, reaching lengths of up to 2.44 m (Klauber Citation1997). Due to its extreme size and consequent large venom yield, C. adamanteus is arguably the most dangerous snake species in the United States and is one of the major sources of snakebite mortality throughout its range that was considerably larger and less patchy (Gold et al. Citation2002).
In this study, an adult female specimen of C. adamanteus was collected from Clermont, Florida (28°55′N, 71°70′W). Genomic DNA was extracted from muscle tissues using QIAGEN DNEasy Extraction Kit following the manufactures instructions. The isolated DNA was stored in the sequencing company (HuiTong Tech, Shenzhen, China). Purified DNA was fragmented and used to construct the sequencing libraries following the instructions of NEBNext® Ultra™ II DNA Library Prep Kit (NEB, BJ, CN). Whole genomic sequencing was performed by the Illumina HiSeq 2500 Sequencing Platform (Illumina, CA). Adapters and low-quality reads were removed using the NGS QC Toolkit (Patel and Jain Citation2012). Then assembly as implemented by SPAdes version 3.9.0 (Bankevich et al. Citation2012). Circularization of this mt genome was confirmed using MITObim version 1.9 (Hahn et al. Citation2013). The complete sequence was primarily annotated by ORF prediction in Unipro UGENE (Okonechnikov et al. Citation2012) combined with manual correction. All tRNAs were confirmed using the tRNA scan-SE search server (Lowe and Eddy Citation1997). Other protein-coding genes were verified by BLAST search on the NCBI website (http://blast.ncbi.nlm.nih.gov/), and manual correction for start and stop codons were conducted. This complete mitochondrial genome sequence together with gene annotations was submitted to GenBank under the accession numbers of MH626511.
The complete mitochondrial genome of C. adamanteus was 17,406 bp in length and has a base composition of A (32.3%), T (24.7%), C (29.7%), G (13.3%), demonstrating a bias of higher AT content (57.0%) than GC content (43.0%). The mitochondrial genome contains a typically conserved structure among Crotalinae mitogenomes, encoding 13 protein-coding genes (PCGs), 22 transfer RNA genes (tRNA), 2 ribosomal RNA genes (12S rRNA and 16S rRNA) and a control region (D-loop region). Twelve PCGs were located on the H-strand, except ND6 that was located on the L-strand. ATP8 gene and ATP6 gene were overlapped by 9 bp.
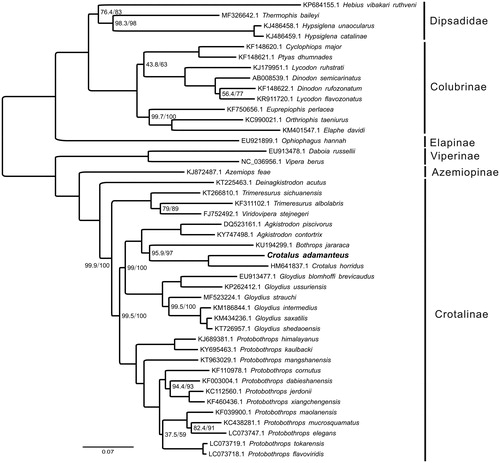
For phylogenetic analysis assessing the relationship of this mitogenome, we selected other 43 Viperidae mitogenomes from Dipsadidae (4 taxa), Colubrinae (9 taxa), Elapinae (1 taxon), Viperinae (2 taxa), Azemiopinae (1 taxon), and Crotalinae (26 taxa) to construct genome-wide alignment. The genome-wide alignment of all mt genomes was done by HomBlocks (Bi et al. Citation2018) under trimAl method, which containing all phylogenetic informative gaps in alignments, resulting in 17,595 positions in total, including almost all PCGs and rRNA genes. The whole genome alignment was analysed by IQ-TREE version 1.6.6 (Nguyen et al. Citation2015) under the GTR + F+R6 model. The tree topology was verified under both 1000 bootstrap and 1000 replicates of SH-aLRT test. The resulting tree was represented and edited using FigTree version 1.4.1 (Rambaut and Drummond Citation2012). As shown in , the phylogenetic positions of these 44 mt genomes were successfully resolved with high bootstrap and SH-aLRT supports across almost all nodes. The result indicated C. adamanteus clustered with Crotalus horridus (HM641837.1), representing a distinct genus, which has the closest relationship with Bothrops jararaca (KU194299.1).
Figure 1. Phylogenetic tree yielded by IQ-TREE of 44 Viperidae mitogenomes. Consensus tree is shown with support indicated by numbers at branches, representing percentages of SH-aLRT test, and bootstraps. Fully phylogenetical resolved nodes are not labelled.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19:455–477.
- Bi G, Mao Y, Xing Q, Cao M. 2018. HomBlocks: a multiple-alignment construction pipeline for organelle phylogenomics based on locally collinear block searching. Genomics. 110:18–22.
- Gold BS, Dart RC, Barish RA. 2002. Bites of venomous snakes. N Engl J Med. 347:347–356.
- Hahn C, Bachmann L, Chevreux B. 2013. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads—a baiting and iterative mapping approach. Nucleic Acids Res. 41:e129.
- Klauber LM. 1997. Rattlesnakes: their habits, life histories, and influence on mankind. 2nd ed. Berkeley (CA): University of California Press.
- Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25:955–964.
- Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32:268–274.
- Okonechnikov K, Golosova O, Fursov M. 2012. Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics. 28:1166–1167.
- Patel RK, Jain M. 2012. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS One. 7:e30619.
- Rambout A, Drummond A. 2012. FigTree: tree figure drawing tool, v1. 4.1. Institute of Evolutionary Biology, University of Edinburgh.