
Abstract
Prunus incisa Thunb is a rare Prunoideae species distributed in the Fuji region, Japan. Here, we assembled the complete chloroplast (cp) genome of P. incisa based on Illumina sequencing. The cp genome of P. incisa was 157,943 bp in length with 129 genes comprising 84 protein-coding genes, 37 tRNA genes and 8 rRNA ribosomal genes. The overall GC content of cp genome was 36.7%. A maximum likelihood phylogenetic analysis revealed that P. incisa was sister to P. serrulata.
Prunus incisa Thunb (Rosaceae), also known as Fuji cherry, is a deciduous tree endemic to the Fuji region. The species is a valuable ornamental plant with low height of 2 m to 5 m and various flower colours from white to rose, which is always found as shrub than a small tree (Ohwi J et al. Citation1966). It can be used for hedging and potted up for bonsai. Besides, the species is often applied for cross breeding as excellent parent which yields improved cultivar ‘Umineko’, ‘Semperflorens’, ‘Fuyu-zakura’ etc. Here, we assembled the chloroplast (cp) genome of P. incisa and analyzed its phylogenetic status in Prunoideae which will be helpful for future studies on the conservation, phylogeny, and breeding of Prunus.
We collected fresh leaves of P. incisa from Wuhan Faya Garden Group Co., Ltd., Hubei province, China (30°29′14.8″N 114°33′1.7″E). The leaves were stored in the refrigerator at −80 °C. The genomic DNA extracted from a single individual of P. incisa was used to generate a short-insert (<800 bp) paired-end sequencing library and sequenced using a HiSeq XTM Ten analyzer (Illumina, San Diego, CA, USA) at Beijing Genomics Institute (BGI, Shenzhen, China). The chloroplast DNA sequences were manually confirmed and assembled using Sequencher software (Gene Codes Corporation, Ann Arbor, MI, USA) (v5.4) and Geneious software (Biomatters Ltd., Auckland, New Zealand ) (R10.2.3) software, and cp genome annotation was performed using the Dual Organellar Genome Annotator (DOGMA; Wyman et al. Citation2004).
The complete chloroplast genome of P. incisa was 157,943 bp in length and contained a pair of inverted repeat (IR, 26,436 bp) regions, which were separated by a small single copy (SSC, 19,143 bp) region and a large single copy (LSC, 85,928 bp) region. The whole cp genome encoded 112 unique genes, of which 18 were duplicated in the IRs, giving a total of 129 genes, including 84 protein-coding genes (PCG), 37 tRNA genes, eight rRNA ribosomal genes. Of these genes, 15 genes contained one intron, while two genes had two introns. The overall GC content of P. incisa cp genome was 36.7% with 34.6, 30.2, and 42.5% GC in the LSC, SSC, and IR regions, respectively.
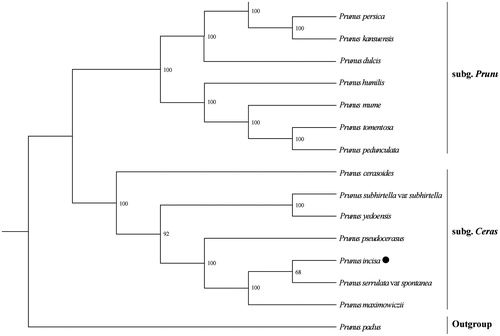
The complete cp genome sequence of P. incisa was used to construct the phylogenetic tree with 16 Prunoideae species of which cp genome sequences were downloaded from GenBank database. The maximum likelihood (ML) was performed using RAxML-HPC2 v8.2.10 (Stamatakis, Citation2014) with 1000 bootstrap replicates on the CIPRES Science Gateway v. 3.3 (Miller et al., Citation2010). The ML tree showed that acoll the species were divided into two clades, subg. Prunus and subg. Cerasus, which is consistent with previous results (Shi et al., Citation2013), and P. incisa was sister to P. serrulata ().
Figure 1. Maximum likelihood phylogenetic tree for Prunus incisa based on 16 complete cp genomes. The accession numbers are listed as below: Prunus mongolica (MG602256); P. persica (HQ336405); P. kansuensis (NC_023956); P. dulcis (NC_034696); P. humilis (NC_035880); P. mume (NC_023798); P. tomentosa (NC_036394); P. pedunculata (MG602257); P. cerasoides (NC_035891); P. subhirtella var subhirtella (KP760075); P. yedoensis (KP732472); P. pseudocerasus (NC_030599); P. serrulata var spontanea (KP760073); P. maximowiczii (KP760071); P. padus (KP760072).
Acknowledgments
We would like to thank Wuhan Faya Garden Group Co., Ltd. for providing fresh leaves of P. incisa.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Miller MA, Pfeiffer W, Schwartz T. 2010. Creating the CIPRES Science Gateway for Inference of Large Phylogenetic Trees. New Orleans, LA: Gateway Computing Environments Workshop (GCE).
- Ohwi J, Meyer FG, Walker EH. 1966. Flora of Japan. IIb:132.
- Shi S, Li J, Sun J, Yu J, Zhou S. 2013. Phylogeny and Classification of Prunus sensu lato (Rosaceae). J Integr Plant Biol. 55:1069–1079.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30:1312–1313.
- Wyman SK, Jansen RK, Boore JL. 2004. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 20:3252–3255.