
Abstract
The complete mitogenome of Echinometra sp. EZ has been described and fully annotated in this study. Phylogenetic analysis of cytochrome c oxidase subunit I (COI) from six Echinometra species confirms that our sample is E. sp. EZ. The mitogenome is 15,698 bp in length and contains 13 protein-coding genes, 22 tRNAs, 2 rRNAs, and a non-coding region with an identical organization to other Echinoidea. The E. sp. EZ mitogenome shared ∼99.1% identity to the published Echinometra mathaei mitogenome, differing by 147 SNPs. The E. sp. EZ mitogenome will serve as a resource that can be applied to disentangling the Echinometra species complex and to future population genetic studies of this ecologically important sea urchin species.
Introduction
Sea urchins belonging to the genus Echinometra have been widely studied and recognized for their unique patterns of genetic structure and speciation (Mcartney et al. Citation2000; Lessios Citation2006; Bronstein and Loya Citation2013). Echinometra is a pantropical genus with a distribution across the Indo-Pacific, Caribbean, and Atlantic, and plays an important role in coral reef ecosystems as a major bioeroder (Moulin et al. Citation2015). Echinometra species present in the Indo-Pacific region are best understood as a complex consisting of four distinct species. Echinometra mathaei and E. oblonga correspond to types ‘B’ and ‘D,’ respectively (Arakaki and Uehara Citation1999; Mita et al. Citation2004). The other two species are referred to as E. sp. A and E. sp. C (Palumbi et al. Citation1997). A recent study suggests a potential new species or subspecies that occurs in the Gulf of Aqaba/Eilat and reefs surrounding Zanzibar, referred to as E. sp. EZ (Bronstein and Loya Citation2013). Genetic and comparative genomic studies on this species complex will be valuable for answering fundamental biological questions about sympatric speciation, range expansion, and adaptive potential. A complete mitogenome is an informative reference for future studies on understanding the history of this species complex and future genetic studies of hybridization.
Total genomic DNA was extracted from a single gonadal tissue sample of E. sp. EZ collected from the Dhabiya reef site in the Persian/Arabian Gulf (24°21′55.8″N 54°06′02.9″E) with the DNeasy Blood and Tissue Kit (QIAGEN ). High-quality DNA was submitted for PCR-free library preparation and whole genome sequencing on an Illumina HiSeq3000 (100 bp paired-end reads) and a NextSeq500 (100 bp paired-end reads) at the University of Florida Interdisciplinary Center for Biotechnology Research. Trimmomatic v0.38 (Bolger et al. Citation2014) was used for adapter removal and quality filtering. Platanus v1.2.1 (Kajitani et al. Citation2014) was used to assemble reads into contigs and scaffolds with default parameters and K-mer ranging from 49 to 99. From the first pass genome assembly, we successfully identified a complete mitochondrial chromosome using the published E. mathaei mitogenome sequence (accession number: NC_034767.1). Annotations of the mitogenome were computed using Geneious v11.1.4 (Kearse et al. Citation2012) with the published E. mathaei mitogenome sequence as a reference. Given the complex phylogenetics of this species complex, we used BLASTn searches at the National Center for Biotechnology Information (NCBI) with cytochrome c oxidase subunit I gene (COI) to confirm that our sample matched E. sp. EZ. Phylogenetic analysis of eight Echinometra COI sequences (derived from six Echinometra species), suggests that the ‘E. mathaei’ mitogenome previously deposited at NCBI would more accurately be defined taxonomically as E. sp. EZ (Sequence data and maximum likelihood tree available from the Dryad Digital Repository: https://doi.org/10.5061/dryad.20k3fj0).
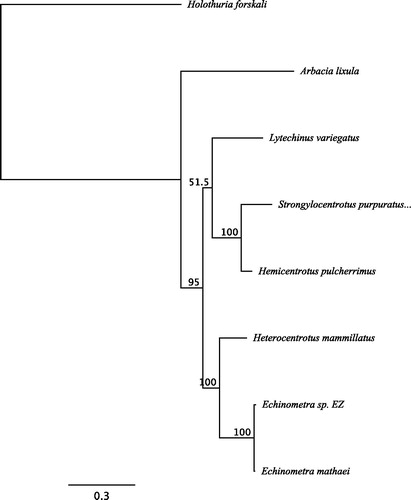
The mitogenome of E. sp. EZ is 15,698 bp in length and contains 13 protein-coding genes, 22 tRNAs, 2 rRNAs, and a non-coding region with an identical organization to other Echinoidea (accession number: MH685644). The mitogenome generated here was aligned to the published E. mathaei genome using the MAFFT alignment with the E-INS-i algorithm implemented in Geneious v11.1.4. We found 147 SNPs differentiating the two individuals (∼99.1% identity) (SNP table available on the Dryad Digital Repository: https://doi.org/10.5061/dryad.20k3fj0). Using this same alignment protocol, we generated a 16,520 column nucleotide alignment of the mitogenomes from Echinometra sp. EZ, Echinometra mathaei, Arbacia lixula, Lytechinus variegatus, Strongylocentrotus purpuratus, Hemicentrotus pulcherrimus, Heterocentrotus mammillatus, and the outgroup Holothuria forskali (Holothuroidea) (Alignment data are available on the Dryad Digital Repository: https://doi.org/10.5061/dryad.20k3fj0). With this alignment, we used MEGA v7.0.21 (Kumar et al. Citation2016) to determine that the best fit model of nucleotide substitution was GTR + G. We then generated a maximum likelihood (ML) tree using this model in the PhyML v3.0 plugin (Guindon et al. Citation2010) in Geneious v11.1.4 (with 1000 bootstrap replicates). The ML tree is in agreement with other published phylogenies based on morphological characteristics and mitochondrial regions () (Smith et al. Citation2006; Kroh and Smith Citation2010).
Figure 1: The maximum likelihood (ML) tree generated using Geneious v11.1.4 with six Echinoidea species: Strongylocentrotus purpuratus (accession number: X12631.1), Lytechinus variegatus (NC_037785.1), Heterocentrotus mammillatus (NC_034768.1), Hemicentrotus pulcherrimus (NC_023771.1), Echinometra mathaei (NC_034767.1), Arbacia lixula (NC_001770.1), and one Holothuroidea as an outgroup: Holothuria forskali (FN562582.1). The ML tree was generated with an alignment of the whole mitogenome sequences of all species, using the GTR + G model. The numbers above the branches specify bootstrap percentages (1000 replicates).
Acknowledgments
We thank Edward G. Smith, Grace O. Vaughan, Dain McParland, and Noura Al-Mansoori for help with sample collection.
Disclosure statement
The authors report no conflicts of interest.
Additional information
Funding
Related Research Data
References
- Arakaki Y, Uehara T. 1999. Morphological comparison of black Echinometra individuals among those in the Indo-West Pacific. Zoolog Sci. 16:551–558.
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30:2114–2120.
- Bronstein O, Loya Y. 2013. The taxonomy and phylogeny of Echinometra (Camarodonta: Echinometridae) from the Red Sea and Western Indian Ocean. PLoS ONE. 8:e77374
- Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 59:307–321.
- Kajitani R, Toshimoto K, Noguchi H, Toyoda A, Ogura Y, Okuno M, Yabana M, Harada M, Nagayasu E, Maruyama H, et al. 2014. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 24:1384–1395.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28:1647–1649.
- Kroh A, Smith AB. 2010. The phylogeny and classification of post-palaeozoic echinoids. J Syst Palaeontol. 8:147–212.
- Kumar S, Stecher G, Tamura K. 2016. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 33:1870–1874.
- Lessios H. 2006. Speciation in sea urchins. Echinoderms. Durham Proceedings of the 12th International Echinoderm Conference 91–101.
- McCartney MA, Keller G, Lessios HA. 2000. Dispersal barriers in tropical oceans and speciation in Atlantic and eastern Pacific sea urchins of the genus Echinometra. Mol. Ecol. 9:1391–1400.
- Mita M, Uehara T, Nakamura M. 2004. Speciation in four closely related species of sea urchins (genus Echinometra) with special reference to the acrosome reaction. Invertebr Repr Dev. 45:169–174.
- Moulin L, Grosjean P, Leblud J, Batigny A, Collard M, Dubois P. 2015. Long-term mesocosms study of the effects of ocean acidification on growth and physiology of the sea urchin Echinometra mathaei. Marine Environ Res. 103:103–114.
- Palumbi SR, Grabowsky G, Duda T, Geyer L, Tachino N. 1997. Speciation and population genetic structure in tropical Pacific sea urchins. Evolution. 51:1506–1517.
- Smith AB, Pisani D, Mackenzie-Dodds JA, Stockley B, Webster BL, Littlewood TJ. 2006. Testing the molecular clock: molecular and paleontological estimates of divergence times in the Echinoidea (Echinodermata). Mol Biol Evol. 23:1832–1851.