
Abstract
We determined two complete mitochondrial sequences of female-transmitted (F) mitogenomes of two Hyriopsis schlegelii specimens from this species’ original habitat, Lake Biwa. The mitogenomes were both 15,951 bp in length, and the gene contents and orders agreed with those of the typical F mitogenome of Hyriopsis. Molecular phylogenetic analysis based on the previously identified 13 partial sequences confirmed that the two mitogenomes both belonged to H. schlegelii and not to a closely related Chinese species, Hyriopsis cumingii. The same analysis revealed that two mitogenomes (HQ641406 and FJ529186) previously published as H. schlegelii and H. cumingii might be misidentified.
Hyriopsis schlegelii is a freshwater pearl-producing bivalve originally endemic to Lake Biwa in central Japan. This species was known worldwide as the Biwa pearl mussel but is now threatened in its original habitat and classed as a critically endangered species (CR + EN) in the Japanese Red Data Book (Ministry of the Environment Citation2005). One probable threat to wild H. schlegelii populations is genetic contamination from the closely related Chinese species Hyriopsis cumingii (Matsuda Citation2016) although this theory has not yet been confirmed by a genetic survey. Genetic differences between the two species were clarified via partial sequencing of the COX2-COX1 region of mitochondrial (mt) DNA and partial ITS1 region sequencing of nuclear DNA by Shirai et al. (Citation2010) in their molecular phylogenetic study, which primarily used 20 specimens collected from Lake Biwa before the introduction of H. cumingii. However, the whole mitochondrial genome (mitogenome) sequence of H. schlegelii in its native habitat has not previously been reported.
In this study, we determined the mitogenome sequences of two specimens of Hyriopsis mussel from a pearl culture farm in Lake Biwa. The specimens were deposited at Lake Biwa Museum, Shiga Prefecture, Japan, under registration numbers LBM-1300014556 and LBM-1300014557. Genomic DNA was isolated from the specimens’ foot muscle tissue and sequenced using an Illumina MiSeq (Illumina). The resultant reads were assembled using CLC Genomic Workbench (ver. 11.01; QIAGEN). Contigs were annotated by alignment with a female-transmitted (F) mitogenome sequence published as H. cumingii (FJ529186). Using the four available F mitogenomes of Hyriopsis (including the two sequenced here) and the F mtDNA sequences of Hyriopsis published in Shirai et al. (Citation2010), we constructed a phylogenetic tree using a supermatrix approach (de Queiroz and Gatesy Citation2007); the method of tree construction is described in the legend of .
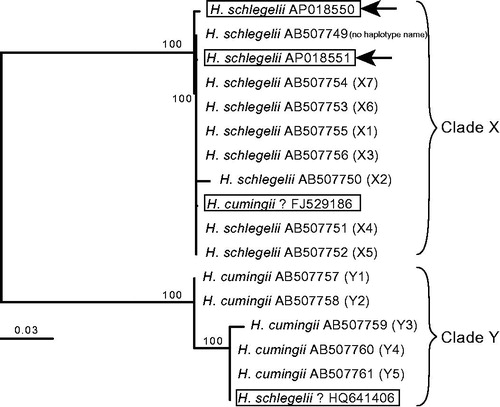
Figure 1. Supermatrix tree of four whole mitochondrial (15,939–15,954 bp) and 13 partial COX2-COX1 region (1027 bp) sequences of female-transmitted (F) mitogenomes of Hyriopsis schlegelii and H. cumingii. Bootstrap support (80% or over) is indicated at the nodes. Accession numbers are indicated after the species names with haplotype names of Shirai et al. (Citation2010) in parentheses for partial sequences. Species names and accession numbers of the four mitogenomes are boxed, and the two mitogenomes sequenced in this study are indicated by arrows. The tree backbone was first generated for the four mitogenomes by the neighbor-joining (NJ) method using the online version of MAFFT (https://mafft.cbrc.jp/alignment/server/). The obtained NJ tree was then used as a backbone constraint for the supermatrix tree. The supermatrix tree was constructed based on the dataset including the four mitogenome and 13 partial sequences, which were first multiple aligned using MAFFT and corrected by eye using Mesquite (version 3.31; http://www.mesquiteproject.org). For the resultant 16,003-bp dataset, maximum likelihood analysis was performed using RAxML BlackBox (https://embnet.vital-it.ch/raxml-bb/).
The mitogenome sequences obtained (DDBJ accession nos. AP018550 and AP018551) were both 15,951 bp in length. Male (M) and female (F)-transmitted mitogenomes in freshwater mussels are known to have different gene orders (Breton et al. Citation2009). The gene order of our mitogenomes differed from that of the H. cumingii M mitogenome (KC150028: 17,100 bp) but was identical to this species’ F mitogenome (FJ529186: 15,954 bp), which confirmed that the newly obtained sequences were those of the F mitogenome.
The resultant supermatrix tree recovered two major clades of the tree published in Shirai et al. (Citation2010): clade X corresponds to H. schlegelii and clade Y to H. cumingii (). Our two mitogenomes were both nested within clade X, which confirmed they came from H. schlegelii. These two sequences are the first F mitogenome sequences of H. schlegelii reported from its native habitat. Based on their phylogenetic positions in the tree, however, the two remaining mitogenomes, FJ529186 (published as H. cumingii) and HQ641406 (published as H. schlegelii), seem to have been misidentified. These possibly misidentified sequences were reported from China, where the native H. cumingii has been hybridized with the introduced H. schlegelii for selective breeding (Peng et al. Citation2014).
Geolocation information
35°05’40.34”N 135°56’51.01”E
Acknowledgments
We thank Masanari Matsuda (Lake Biwa Museum) for registering our specimens in the LBM shell collection. Our special thanks go to Fumio Mikami and Toshiaki Ibuki for help obtaining the specimens.
Disclosure statement
The authors report no conflict of interest. The authors alone are responsible for the content and writing of this manuscript.
Additional information
Funding
Related Research Data
References
- Breton S, Beaupré HD, Stewart DT, Piontkivska H, Karmakar M, Bogan AE, Blier PU, Hoeh WR. 2009. Comparative mitochondrial genomics of freshwater mussels (Bivalvia: Unionoida) with doubly uniparental inheritance of mtDNA: gender-specific open reading frames and putative origins of replication. Genetics. 183:1575–1589.
- de Queiroz A, Gatesy J. 2007. The supermatrix approach to systematics. Trends Ecol Evol (Amst.). 22:34–41.
- Matsuda M. 2016. Hyriopsis schlegeli. In: Scientific Committee for Research into the Wildlife in Shiga Prefecture, editor. Red Data Book of Shiga Prefecture 2015. Hikone: Sunrise Press; p. 578. Japanese
- Ministry of the Environment, editor. 2005. Threatened Wildlife of Japan, Red Data Book. 2nd ed. Vol. 6, Land and Freshwater Mollusks. Tokyo, Japan Wildlife Research Center. Japanese with English summary
- Peng K, Wang CY, Wang JH, Sheng JQ, Shi JW, Li J, Hong YJ. 2014. Molecular cloning, sequence analysis, and cadmium stress-rated expression changes of BTG1 in freshwater pearl mussel (Hyriopsis schlegelii). Zool Res. 35:389–397.
- Shirai A, Kondo T, Kajita T. 2010. Molecular markers reveal genetic contamination of endangered freshwater pearl mussels in pearl culture farms in Japan. Venus. 68:151–163.