
Abstract
Two complete and one near-complete mitochondrial genomes (mitogenomes) were determined for a deep-sea holothurian species of the genus Scotoplanes (Elpidiidae). Each sequence contains two ribosomal RNA genes, 22 transfer RNA genes, and 13 protein-coding genes, as found in most previously determined holothurian mitogenomes. The protein-coding genes use ATG or ATA as the start codon, and TAA or TAG as the stop codon. Phylogenetic reconstruction using the maximum likelihood method confirmed the sister relationship between Scotoplanes sp. and another elpidiid holothurian Peniagone sp.; the three Scotoplanes sp. specimens were recovered as a monophyletic group.
Holothurians of the family Elpidiidae (Elasipodida) are dominant deposit feeders in deep-sea communities (Gutt and Piepenburg Citation1991; Taylor et al. Citation2014). Associations with parasitic gastropods and tanaids (Alvaro et al. Citation2011; Takano et al. Citation2018), and mutualistic decapods (Barry et al. Citation2017) suggest their ecological importance. Species of the elpidiid genus Scotoplanes (or ‘sea pig’) are widely distributed, with S. globosa inhabiting the bathyal and abyssal depths of various sea areas (Gutt and Piepenburg Citation1991; Thandar Citation1999). Ongoing molecular and morphological studies have detected a cryptic species of S. globosa in the northeast Pacific (Barry et al. Citation2017). Molecular comparisons are required to improve our understanding of the diversity and evolutionary history of Scotoplanes species. Here, we sequenced the mitochondrial genomes (mitogenomes) of S. globosa-like specimens from off the coast of Japan in the northwest Pacific; hereafter the study material is referred to as Scotoplanes sp. because the sampling site is quite distant from the type locality of S. globosa (Théel Citation1879) and it might be a cryptic species.
One beam-trawl haul at station OT6 of the R/V Shinsei-maru cruise KS-16-18 (off Otsuchi, Iwate, Japan: 39°18′20″–19′″N, 142°49′48″54″E) yielded hundreds of Scotoplanes sp. from the muddy bottom (1672–1692 m depth; 2.2 °C). Three Scotoplanes sp. specimens (H4, H5 and H8) were preserved in pure ethanol; the sequences of short DNA fragments from the first two specimens were reported previously (Takano et al. Citation2018). Voucher material has been deposited at the Atmosphere and Ocean Research Institute, The University of Tokyo, Japan. Total DNA was extracted from the epidermis of each specimen by using a QIAGEN Genomic-tip 500/G. Paired-end sequencing (2 × 150 bp) was performed in an Illumina NextSeq sequencer. Bases with a quality score lower than 20 were removed from the sequence reads, and trimmed reads shorter than 127 bp were discarded by using Sickle 1.33 (Joshi and Fass Citation2011). The remaining reads were assembled using SPAdes 3.10.1 (Bankevich et al. Citation2012). Gaps in the assembled sequences were filled by Sanger-sequencing with specific primers. Finally, two complete mitogenomes (H4 and H8, DDBJ/EMBL/GenBank accession nos LC416624 and LC416626, respectively) and one near-complete mitogenome (H5, LC416625) were obtained. The assembled sequences were annotated by using the MITOS WebServer (Bernt et al. Citation2013); some annotations were corrected by reference to previously determined holothurian mitogenomes.
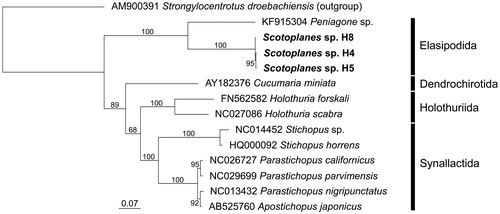
The complete Scotoplanes sp. mitogenome sequences were 15,913 bp (H4) and 15,910 bp (H8) in length, varying by the length of the noncoding region between genes encoding transfer RNAs (tRNAs) tRNACys and tRNAGln. All three mitogenomes obtained here contained two ribosomal RNA (12S and 16S) genes, 22 tRNA genes, and 13 protein-coding genes (PCGs). The length and intraspecies variation of amino acid sequences, and the start and stop codons of each PCG are summarized in Table S1 (available on figshare: https://figshare.com/s/2c8c2a9334ea11ddae39, DOI: 10.6084/m9.figshare.7039331). By partitioned maximum likelihood analysis, the three Scotoplanes sp. specimens were recovered as a well-supported clade (bootstrap percentage [BP] = 100%; ), and a sister relationship between Scotoplanes sp. and another elpidiid species Peniagone sp. was observed (BP = 100%).
Figure 1. Maximum likelihood (ML) phylogeny of 13 holothurians based on concatenated amino acid sequences of 13 protein-coding genes (3570 positions). Scotoplanes sp. is shown in bold. Also included in the analysis was the echinoid Strongylocentrotus droebachiensis as an outgroup taxon, according to relationships among the five echinoderm classes inferred from phylogenomic analysis (Telford et al. Citation2014). Sequences were aligned separately for each gene by using MAFFT 7.397 (Katoh and Standley Citation2013) with default parameters. Ambiguously aligned positions were removed by Gblocks 0.91b (Castresana Citation2000); the ‘Allowed gap positions’ was set to ‘With half'. ML analysis was performed in RAxML 8.2.10 (Stamatakis Citation2014) using the mtREV + G model. Numbers above or below branches denote bootstrap percentages (1000 replicates). DDBJ/EMBL/Genbank accession numbers are shown for published sequences.
Acknowledgements
We are grateful to the captain, crew, and researchers of the R/V Shinsei-maru cruise KS-16-18, which is a part of the research project Tohoku Ecosystem-Associated Marine Sciences funded by the Ministry of Education, Culture, Sports, Science, and Technology, Japan. Sequence assembly was conducted by the Bioengineering Lab. Co., Ltd., Japan.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Alvaro MC, Błażewicz-Paszkowycz M, Davey N, Schiaparelli S. 2011. Skin-digging tanaids: the unusual parasitic behavior of Exspina typica in Antarctic waters and worldwide deep basins. Antarct Sci. 23:343–348.
- Bankevich A, Nurk S, Antipov D, Gurevich A, Dvorkin M, Kulikov AS, Lesin V, Nikolenko S, Pham S, Prjibelski A, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19:455–477.
- Barry JP, Taylor JR, Kuhnz LA, De Vogelaere AP. 2017. Symbiosis between the holothurian Scotoplanes sp. A and the lithodid crab Neolithodes diomedeae on a featureless bathyal sediment plain. Mar Ecol. 38:e12396.
- Bernt A, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69:313–319.
- Castresana J. 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 17:540–552.
- Gutt J, Piepenburg D. 1991. Dense aggregations of three deep-sea holothurians in the southern Weddell Sea, Antarctica. Mar Ecol Prog Ser. 68:277–285.
- Joshi NA, Fass JN. 2011. Sickle: a sliding-window, adaptive, quality-based trimming tool for FastQ files (version 1.33) [Software]; [accessed 2018 Sep 3] https://github.com/najoshi/sickle.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30:772–780.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30:1312–1313.
- Takano T, Itoh H, Kano Y. 2018. DNA-based identification of an echinoderm host for a deep-sea parasitic snail (Gastropoda: Eulimidae). Molluscan Res. 38:212–217.
- Taylor JR, De Vogelaere AP, Burton EJ, Frey O, Lundsten L, Kuhnz LA, Whaling PJ, Lovera C, Buck KR, Barry JP. 2014. Deep-sea faunal communities associated with a lost intermodal shipping container in the Monterey Bay National Marine Sanctuary, CA. Mar Pollut Bull. 83:92–106.
- Telford MJ, Lowe CJ, Cameron CB, Ortega-Martinez O, Aronowicz J, Oliveri P, Copley RR. 2014. Phylogenomic analysis of echinoderm class relationships supports Asterozoa. Proc R Soc B. 281:20140479.
- Thandar AS. 1999. Deep-sea holothuroids taken by the R.V. Africana II in 1959, from of the west coast of the Cape Peninsula, South Africa. Ann S Afr Mus. 105:363–409.
- Théel H. 1879. Preliminary report of the Holothuridae of the exploring voyage of H. M. S. “Challenger”, under Professor Sir C. Wyville Thomson F. R. S., Part 1. Bihang till K Svenska Vet Akad Handlinger. 5:1–20.