
Abstract
The first complete ancient mitochondrial genome of a vicuña (Vicugna vicugna) (3200–2400 B.P.) recovered from the Tulán-54 site (San Pedro de Atacama, Northern of Chile) was sequenced using mitochondrial enrichment by hybridization capture and subsequent Next Generation Sequencing (NGS). Morphometric analyses place this individual inside of a small size camelid group and Bayesian phylogenetic analysis confirms that this specimen belonged to an ancient vicuña.
Four South American camelids are living along the Andes nowadays, two wild species, guanaco (Lama guanicoe) and vicuña (Vicugna vicugna) and two domestic species, llama (Lama glama) and alpaca (Vicugna pacos). Morphometric analyses based on phalanx and astragalus measures can only separate species in two size groups, large size (guanaco and llama) and small size (vicuña and alpaca); thus the identification of wild and domestic species intra size group is very challenging (Cartajena Citation2009). We recovered a camelid bone sample (C20 TULÁN-54) from Tulán-54 (Early Formative period, 3200–2300 years B.P), an archaeological site located in Northern Chile (–23,7629 Latitude and –68.1324 E longitude).
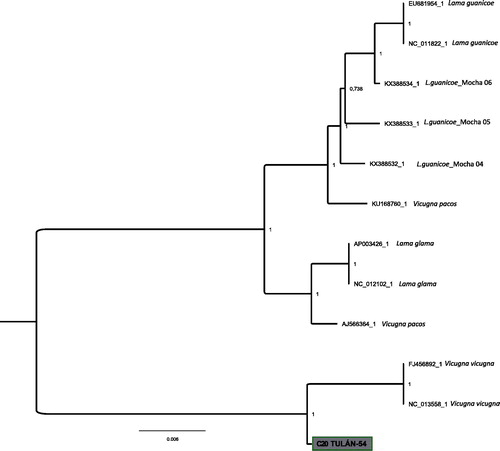
Previous to DNA extraction, morphometric measures were taken on the astragalus bone selected for analysis. Osteometry grouped this specimen as small size camelid. DNA was isolated in a dedicated ancient DNA laboratory at the Centre for GeoGenetics, (University of Copenhagen, Denmark). From this specimen, 150 mg of bone was drilled using a Dremel tool to obtain a bone powder. DNA extractions were performed as in Dabney et al. (Citation2013). Double-stranded libraries were constructed using 21.25 µl of the extracts following the protocol from Meyer and Kircher (Citation2010). Our sequence library was target-captured prior to sequencing to enrich the library for endogenous mitochondrial DNA sequences. The enriched library was quantified using Agilent 2200 TapeStation System, pooled equimolarly, and sequenced on Illumina HiSeq 2500 (80 basepair (bp) single-end) at the Danish National High- Throughput Sequencing Centre. A total of 6,518,814 raw reads were obtained. Raw reads processing (adapter trimming), mapping (read alignment, PCR duplicate removal, indel realignment) and damage analyses were performed using the PALEOMIX pipeline (Schubert et al. Citation2014). The resulting BAM file consisted of 25,688 unique reads and was analyzed using Geneious v7 (Kearse et al. Citation2012) to generate a mitochondrial consensus sequence using the ‘strict’ consensus that calls bases using majority rule and requires a minimum depth-of-coverage of 3×. As a result, the ancient mitogenome of the small size camelid consists of 16,103 bp with an average depth of 71×. A mitogenomic Bayesian phylogeny was created to determine the phylogenetic position of the sample. A total of eight modern mitogenomes were download from NCBI (two alpacas: AJ566364_1, KU168760_1; two vicuñas: FJ456892_1, NC_013558_1; two guanacos: EU681954_1, NC_011822_1 and two llamas: (AP003426_1, NC_012102_1) as well as three ancient guanaco mitogenomes (KX388532_1, KX388533_1 and KX388534_1). The final a1ignment consisted of 16,172 bp. A Markov Chain Monte Carlo (MCMC) was implemented in the software MrBayes v3.2.6 (Ronquist and Huelsenbeck Citation2003) applying a General Time Reversible Model with a proportion of invariable sites and a gamma-shaped distribution. The (MCMC) was run twice with four chains of 1 × 106 generations, sampling every 1 × 103 generations with a 25% burn-in. The resulting phylogenetic tree () shows two big monophyletic clades, one of them only including vicuñas and the ancient sample from Tulán-54, C20, while the other clade consists of domestic camelids, alpacas, llamas and guanacos. Our analysis shows that ancient DNA analysis can shed light onto the identification of camelid species from archaeological sites.
Figure 1. Phylogenetic tree based on complete mitochondrial genomes of modern camelids and one ancient camelid from Tulán-54 (San Pedro de Atacama, Antofagasta Region, Chile). The newly-sequenced specimen is colored in grey.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Cartajena I. 2009. Explorando la variabilidad morfométrica del conjunto de camélidos pequeños durante el Arcaico Tardío y el Formativo Temprano en Quebrada Tulán, norte de Chile. Rev Del Mus Antropol. 2:199–214.
- Dabney J, Knapp M, Glocke I, Gansauge M-T, Weihmann A, Nickel B, Valdiosera C, García N, Pääbo S, Arsuaga J-L, et al. 2013. Complete mitochondrial genome sequence of a Middle Pleistocene cave bear reconstructed from ultrashort DNA fragments. Proc Natl Acad Sci USA. 110:15758–15763.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics [Internet]. [Cited 2018 Sep 13]. 28:1647–1649.
- Meyer M, Kircher M. 2010. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb Protoc. 2010:pdb-prot5448.
- Ronquist F, Huelsenbeck JP. 2003. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 19:1572–1574.
- Schubert M, Ermini L, Sarkissian CD, Jónsson H, Ginolhac A, Schaefer R, Martin MD, Fernández R, Kircher M, McCue M, et al. 2014. Characterization of ancient and modern genomes by SNP detection and phylogenomic and metagenomic analysis using PALEOMIX. Nat Protoc. 9:1056–1082.