
Abstract
In this study, the complete mitochondrial genome of Idgia oculata Redtenbacher, 1868 was sequenced using Illumina’s NovaSeq platform. The mitogenome is a double-stranded circular molecule of 15,805 bp in length with 22 transfer RNA genes, 13 protein-coding genes (PCGs), 2 ribosomal RNA genes as in other insects. To estimate the taxonomic status of Prionoceridae, total of 9 species from 5 families of Cleroidea were selected as ingroups and 2 species of Coccinellidae as outgroups for phylogenetic analysis based on 13PCGs. The results showed that three major clades were formed, including a ‘Melyridae-Cleridae’ clade, ‘Melyridae-Trogossitidae-Phloiophilidae’ clade, and ‘Melyridae-Prionoceridae’ clade. Prionoceridae showed more closely related to Melyridae than other families. More mitogenome of thorough taxon sampling will be needed to well understand the relationship in Cleroidea.
Idgia oculata Redtenbacher, 1868 is a species of the family Prionoceridae within the melyrid lineage (‘soft-winged flower beetles’) of the superfamily Cleroidea (Polyphaga: Cucujiformia) (Bouchard et al. Citation2011). The species is easily recognized by the large-sized body, length 15.5–18.5 mm, metallic blue head and elytra, testaceous pronotum, with a pair of blackish spots on the center of the disc.
Idgia oculata is normally encountered in large groups, feeding on various flowering shrubs and trees. Flight period centered around June with extreme dates of 25 May and 6 July (Aston Citation2011).
The specimens used in this study was collected by a flight interception trap (FIT) (Nie et al. Citation2017) from Hong Kong and deposited in the Institute of Zoology, Chinese Academy of Sciences, Beijing, China. Genomic DNA was extracted by DNeasy Blood & Tissue kit (QIAGEN, Germany) and then sequenced using Illumina’s NovaSeq platform (Illumina, San Diego, CA) with 350 bp insert size and a pair-end 150 bp sequencing strategy. The sequence reads were first filtered by the programs following Zhou et al. (Citation2013) and then the remaining high-quality reads were assembled using IDBA-UD (Yu and Henry Citation2012). The annotations of genes were done by Geneious 8.0.5 software (Kearse et al. Citation2012) and tRNAscan-SE 1.21 (Schattner et al. Citation2005).
The complete mitochondrial genome (mitogenome) of I. oculata is a double-stranded circular molecule of 15,805 bp in length (GenBank accession number: MH779812), with 22 transfer RNA genes, 13 protein-coding genes (PCGs), 2 ribosomal RNA genes, and a control region as in other insects. The overall base composition is A: 40.1%, T: 40.8%, C: 11.2%, and G: 7.9%, with a much higher A + T content.
The phylogenetic tree was reconstructed to estimate the status of Prionoceridae in Cleroidea. All available mitogenomes of families of Cleroidea were downloaded from Genbank ().
Table 1. The information of 11 species used for phylogenetic analysis.
The acceptable sequences including 13 protein-coding genes and longer than 10K bp were kept. Total 8 species (JX412815.1, EU877951.1, JX412799.1, JX412833.1, JX412765.1, KX035157.1, KT808467.1, JX412752.1) from 5 families were selected as ingroups and 2 species (JQ321839.1, KU877170.1) of Coccinellidae was selected as outgroups (). The phylogenetic inference was done based on 13PCGs. Trans Align methods were used to align all protein-coding genes (Bininda-Emonds Citation2005). The aligned data from 13PCGs were concatenated with Sequence Matrix v.1.7.8 (Vaidya et al. Citation2011). Bayesian inference was performed using MrBayes v.3.2 (Ronquist et al. Citation2012). Data were partitioned according to loci of 13 PCGs. The MCMC search was conducted for 2,000,000 generations, and sampling was done every 100 generations until the average standard deviation of split frequencies was below 0.01. The first 25% of trees were discarded as ‘burn-in’ and posterior probabilities were estimated for each node.
Phylogenetic tree () showed that three major clades were formed, including ‘Melyridae-Cleridae’ clade, ‘Melyridae-Trogossitidae-Phloiophilidae’ clade, and ‘Melyridae-Prionoceridae’ clade. Melyridae is distributed on three branches, which suggests it is not recovered as a monophyly but a polyphyly. This result is congruent with the proposal by Majer (Citation1987) that the Melyridae is polyphyletic based on the morphological study.
Figure 1. The Bayesian tree based on 13PCGs combined data sets. Numbers on nodes indicate Bayesian posterior probabilities. Red branch is the new data in this study.
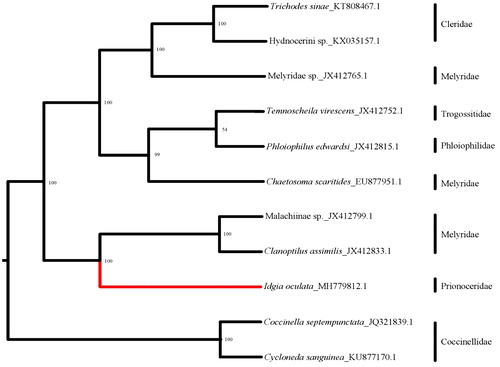
Prionoceridae seems more closely related to the melyrid subfamily Malachiinae than other taxa based on current data. But it was formed a clade with the melyrid subfamily Melyrinae based on morphological characters in the cladograms of all Cleroidea produced by Kolibáč (Citation1999). However, few molecular data affect the further discussion on the basal relationship of Cleroidea. More mitogenome of thorough taxon sampling will be needed to resolve this question, especially the relationship between Prionoceridae and Melyridae.
Acknowledgements
We are grateful to Meixia Yang, Haidong Yang, Tengfei Qiu, Chao Yang, Yongying Ruan, You Li, Xingming Wang, Zhong Peng, and fellows of the Beetle Working Group of Agriculture, Fisheries and Conservation Department (AFCD) of Hong Kong SAR Government for the field work in Hong Kong. We thank Peng Zhang who is from Berry Genomics Corporation, Beijing, China for help with the sequence.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Aston P. 2011. Prionoceridae Lacordaire 1857 of Hong Kong and Guangdong Province, China (Coleoptera: Cleroidea). Hong Kong Entomol Bull. 3:2–6.
- Bininda-Emonds O. 2005. Trans Align: using amino acids to facilitate the multiple alignment of protein-coding DNA sequences. BMC Bioinf. 6:156.
- Bouchard P, Bousquet Y, Davies AE, Alonso-Zarazaga MA, Lawrence JF, Lyal CHC, New AF, Reid CAM, Schmitt M, Ślipiñski SA. 2011. Family-group names in Coleoptera (Insecta). ZooKeys. 88:1–972.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28:1647–1649.
- Kim MJ, Wan X, Kim I. 2012. Complete mitochondrial genome of the seven-spotted lady beetle, Coccinella septempunctata (Coleoptera: Coccinellidae). Mitochondr. DNA. 23(3):179–181.
- Kolibáč J. 1999. Comparative morphology of mandible, epipharynx and alimentary canal in larval and adult Cleroidea (Coleoptera). Acta Mus Morav Sci Bio (Brno). 84:11–69.
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61:539–542.
- Majer K. 1987. Comparative morphology and proposed major taxonomy of the family Melyridae (Insecta, Coleoptera). Pol Pis Entomol. 56:719–859.
- Nie RE, Yang MX, Xue HJ, Yang YR, Tong YJ, Qiu TF, Bai M, Yang XK. 2017. The application and effectiveness of a flight interception trap for insect collecting. Chin J Appl Ent. 54:530–535.
- Schattner P, Brooks AN, Lowe TM. 2005. The tRNA scan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33:686–689.
- Sheffield NC, Song H, Cameron L, Whiting MF. 2008. A comparative analysis of mitochondrial genomes in Coleptera (Athropoda:Insecta) and Genome Descriptions of Six New Beetles. Mol. Biol. Evol. 25(11):2499–2509.
- Vaidya G, Lohman DJ, Meier R. 2011. Sequence Matrix: concatenation softwarefor the fast assembly of multi-gene datasets with character set and codon information. Cladistics. 27:171–180.
- Yu P, Henry CML. 2012. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics. 28:1420–1428.
- Zhou X, Li YY, Liu SL, Yang Q, Su X, Zhou LL, Tang M, Fu RB, Li JG, Huang QF. 2013. Ultra-deep sequencing enables high-fidelity recovery of biodiversity for bulk arthropod samples without PCR amplification. Gigascience. 2:4.