
Abstract
The complete chloroplast (cp) genome sequence of an endangered tree species, Zelkova schneideriana, was first assembled and characterized based on Illumina pair-end sequencing. The cp genome is 158,991 bp in length, with a quantitative structure comprising a pair of inverted repeats (IRs) of 26,427 bp separated by a large single copy (LSC) of 87,424 bp, a small single copy (SSC) of 18,713 bp. A total of 129 genes were annotated including 84 protein-coding genes, 37 tRNA genes, and 8 rRNA genes, respectively. Phylogenetic analysis based on the cp genomes indicates that Z. schneideriana is closely related to genus Ulums in Ulmaceae.
Zelkova schneideriana Hand.-Mazz., a deciduous tree species belongs to family Ulmaceae, is endemic to central and southern China. It is well-popular by people due to its high medicinal, economic, and ornamental values (Fu et al. Citation2013). The high quality timber is used for manufacturing furniture and the color-leaves widely introduced in the landscapes. However, excessive exploration has led to the natural populations decreased sharply. Consequently, Z. schneideriana has been listed as a second-class national key protected wild plant in China (http://www.gov.cn/gongbao/content/2000/content_60072.htm). Up to date, there’s no comprehensive genomic resource were conducted for Z. schneideriana. Therefore, we characterized the complete chloroplast (cp) genome of Z. schneideriana based on de novo sequencing. Our study will provide a valuable plastid genomic information for its conservation genetic and phylogenetic studies.
The individuals of Z. schneideriana were collected from Nanjing Botanical Garden of Mem. Sun Yat-Sen and voucher specimen was deposited in the Herbarium of Institute of Botany, Jiangsu Province and Chinese Academy of Sciences (NAS). The total genomic DNA was isolated from fresh leaves using Plant Genomic DNA Kit DP305 (Tiangen, Beijing) and sequenced on Illumina Hiseq2500 platform (San Diego, CA). The raw data were assembled using NOVOPlasty 2.7.2 (Dierckxsens et al. Citation2017). The cp genome was annotated with GeSeq (https://chlorobox.mpimp-golm.mpg.de/geseq.html) and adjusted start and stop codons manually. tRNA were validated by tRNAscan-SE (Schattner et al. Citation2005).
The cp genome sequence of Z. schneideriana (MK096789) was 158,991 bp in length. It harboured a typical circular and quadripartite structure including two copies of inverted repeat (IRs, 26,427 bp), a large single copy (LSC, 87,424 bp), and a small single copy (SSC, 18,713 bp). A total of 129 genes were identified, containing 84 protein-coding genes (PCGs), 37 transfer RNA genes (tRNA), and 8 ribosomal RNA genes (rRNA).There were 18 genes (7 PCGs, 7 tRNA, and 4 rRNA) repeated in IR regions. The overall GC content of Z. schneideriana cp genome was 35.6% with 42.4, 33.0, and 28.3% in the IRs, LSC and SSC, respectively.
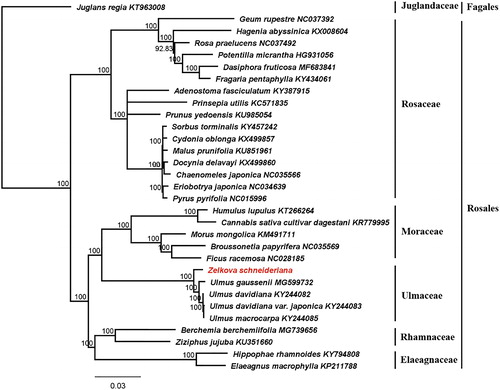
To test the phylogenetic relationship within the Rosales, 30 additional cp genomes were obtained from NCBI. The alignment were generated using the MAFFT 7.409 (Katoh and Standley Citation2013). And molecular phylogenetic tree were reconstructed based on whole cp genome sequences with the maximum likelihood method that employed in RAxML (Stamatakis Citation2014) on the CIPRES gateway (Miller et al. Citation2010). The result reveals that Z. schneideriana is sister to the other species of Ulmus (). We expect that the cp genome of Z. schneideriana will provide valuable data for future studies on conservation genetics, species identification, and breeding of the economically important Zelkova lineage.
Figure 1. Phylogenetic topology of 30 species in Rosales based on cp genomes (the bootstrap values were listed on the branches).
Disclosure statement
The authors declare no conflict of interest.
Additional information
Funding
References
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45:e18.
- Fu L, Xin Y, Alan W. 2013. Flora of China 5: Ulmaceae. Vol. 8. St. Louis: Missouri Botanical Garden Press.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30:772–780.
- Miller MA, Pfeiffer W, Schwartz T. 2010. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. Proceedings of the Gateway Computing Environments Workshop (GCE). IEEE; Nov 14; New Orleans, LA. p. 1–8.
- Schattner P, Brooks A, Lowe T. 2005. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucl Acids Res. 33:W686–W689.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30:1312–1313.