
Abstract
Malus transitoria, belongs to the genus Malus of the family Rosaceae, which is native to China. In this study, our results showed the complete chloroplast genome of M. transitoria 159,847 bp in length consisting of a pair of inverted repeats (IRs) with a length of 26,365 bp, separating by two single-copy regions (LSC, 87,896 bp and SSC, 19,221 bp). In total, 112 genes was annotated comprising of 78 protein-coding genes, 30 tRNA genes, and 4 rRNA genes. A total of 102 SSRs and 49 long repeats were identified in the complete chloroplast genome of M. transitoria. The phylogenetic analysis revealed that M. transitoria has a close relationship with M. yunnanensis.
Malus transitoria, is a crabapple species belong to the genus Malus of the family Rosaceae, main located within Gansu, Inner Mongolia, Qinghai, Shaanxi, Sichuan and Xizang Provinces in China. This species is a type of wild resource plant with the advantage of resistant to drought and cold temperatures, which often serves as a rootstock for the cultivated apples (Editorial Committee of Flora of China, Chinese Academy of Sciences Citation1974). The tender leaves of M. transitoria were processed into tea with a long history, which have good health function, and deeply loved by Tibetan people. Therefore, it has potential application value in the field of breeding, medicine and food. In this article, complete and annotated cp genome of M. transitoria is reported, which has been submitted to GenBank with the accession number MK098838.
Leaf material was collected from Yangling in Shaanxi province (latitude: 34°18′21′′ N, longitude: 108°02′13′′ E; China; voucher AF-06-93 deposited in the Institute of College of Horticulture, Northwest A&F University, Yangling, China). Genomic DNA was extracted from the samples using the DNeasy Plant Mini Kit (Qiagen, Valencia, CA). An Illumina paired-end sequencing library was constructed and generated using the HiSeq X Ten platform. MITObim v1.8 (Hahn et al. Citation2013) software was utilized for de novo genome assembly, with closely related cpDNA sequences of Malus baccata (KX499859) as the reference. The genes were annotated using the program online software DOGMA (Wyman et al. Citation2004). Perl script MISA (Thiel et al. Citation2003) was used to detect single sequence repeats (SSRs) with minimal repeat numbers of 10, 5, 4, 3, 3, and 3 for mono-, di-, tri-, tetra-, penta-, and hexa-nucleotides, respectively. REPuter (Kurtz et al. Citation2001) was used to identify tandem repeats with minimal size was 30 bp and 90% sequence, including forward, palindromic, reverse, and complement repeats.
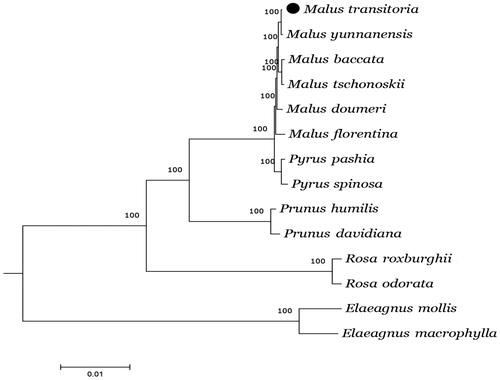
The complete chloroplast genome of M. transitoria is 159,847 bp in size and has a typical quadripartite structure, consisting of a large single copy (LSC) region of 87,896 bp, a small single copy (SSC) region of 19,221 bp, and two inverted repeat regions (IRa and IRb) of 26,365 bp each, of which total CDS length is 112,099 bp and intergenic spacer length is 47,748 bp. The GC content is 36.6%, with the IR regions having higher GC content (42.7%) than the LSC (34.3%) and SSC (30.5%) regions. The genome contains 112 unique genes, including 78 protein-coding genes (PCGs), 30 transfer RNAs (tRNAs), and four ribosomal RNAs (rRNAs). In the IR regions, a total of 19 genes were found duplicated, including eight PCG species, seven tRNA species, and four rRNA species. In the M. transitoria chloroplast genome, 102 SSRs were detected, among which 78 (76.5%) are mono-repeats; 18 (17.6%) are di-repeats; five (4.9%) are tetra-repeats; and one (1.0%) is hexanucleotides repeats. A total of 49 long repeats were identified, comprising 22 forward, 16 palindromic, 10 reverse, and 1 complement repeats. Neighbour-joining (NJ) phylogenetic tree was constructed using MEGA7 with 1000 bootstrap replicate (Kumar et al. Citation2016). As shown in the phylogenetic tree (), M. transitoria was most closely related to M. yunnanensis.
Figure 1. Phylogenetic relationships of 12 sequences in Rosaceae with the out-group of two Elaeagnus species constructed by whole chloroplast genome with the neighbor-joining (NJ) analyses. Numbers in the nodes are the bootstrap values from 1000 replicates. Accession numbers: Rosa roxburghii KX768420, Rosa odorata KF753637, Pyrus pashia KY626169, Pyrus spinosa HG737342, Prunus humilis MF405921, Prunus davidiana MH460864, Malus baccata KX499859, Malus doumeri KX499861, Malus florentina KX499862, Malus tschonoskii KX499863, Malus transitoria MK098838, Malus yunnanensis MH394388, Elaeagnus mollis KY511611, and Elaeagnus macrophylla KP211788.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Editorial Committee of Flora of China, Chinese Academy of Sciences. 1974. Flora of China. Vol. 36. Beijing: Science Press. p. 393–394.
- Hahn C, Bachmann L, Chevreux B. 2013. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads—a baiting and iterative mapping approach. Nucl Acids Res. 41:e129–e129.
- Kumar S, Stecher G, Tamura K. 2016. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol. 33:1870–1874.
- Kurtz S, Choudhuri JV, Ohlebusch E, Schleiermacher C, Stoye J, Giegerich R. 2001. REPuter: the manifold applications of repeat analysis on a genomic scale. Nucl Acids Res. 29:4633–4642.
- Thiel T, Michalek W, Varshney RK, Graner A. 2003. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet. 106:411–422.
- Wyman SK, Jansen RK, Boore JL. 2004. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 20:3252–3255.