
Abstract
In this paper, we report the genomic architecture of a complete mitogenome from a Wedge-tailed eagle (Aquila audax). The mitogenome sequence was circular, and 17,494 bp in length. Compared to other members of the family Accipitridae, the genome encoded a conserved structure consisting of 13 protein-coding genes (PCGs), two rRNA genes, 22 tRNA genes, a repeat region and a control region (D-loop), with all protein-coding sequences started with methionine. The lengths of 12S and 16S ribosomal RNA were 971 bp and 1545 bp, respectively, and were located between tRNA-Phe and tRNA-Leu. The overall base composition of the mitogenome of A. audax was dominated by higher AT (54.3%) than GC (45.7%) content. The complete mitogenome sequence determined in this study would be useful to track the deeper evolutionary history and the conservation of A. audax.
The Wedge-tailed eagle is the largest Australian bird of raptor species and is distributed widely across Australia (Knobel, Citation2015). The estimated population size of the Wedge-tailed eagle is likely to be in the ‘hundreds of thousands’, and the numbers may even be increasing within Australia. At a time when the general trend for many eagle species worldwide is in decline due to direct threats (e.g. poaching and collisions with wind turbines) and indirect threats (e.g. habitat loss and environmental toxins) to populations (Doyle et al. Citation2014), the Wedge-tailed eagle has fared better than most (Doyle et al. Citation2014; Allen and DeCandido Citation2012). Avian mitochondrial genomics lags behind mammalian mitochondrial genomics and there are major uncertainties in the position of many avian species including Wedge-tailed eagle (A. audax) due to a paucity of mitochondrial (mt) datasets. With this in mind, this paper describes the genomic structure of a complete mitogenome from A. audax to facilitate comparative studies of avian genomics and to further strengthen our understanding of the species ecological diversity and host phylogeny for eagle research and conservation.
The blood sample used in this study was sourced from a Wedge-tailed eagle (A. audax) in the wild (year of sampling: 2017; GPS location: 32°16´42.905˝S, 148°34´54.344˝E), was stored in appropriate conditions by the Veterinary Diagnostic Laboratory (VDL), Charles Sturt University under the accession number CS17-0090. Animal sampling was obtained in accordance with approved guidelines set by the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes (1997) and approved by the Charles Sturt University Animal Ethics Committee (Research Authority permit 09/046), and the total genomic DNA was extracted using an established protocol (Das et al. Citation2017; Sarker et al. Citation2016; Sarker et al. Citation2015). The genomic library preparation and sequencing was performed according to the published protocol (Das et al. Citation2017; Sarker et al. Citation2018). Briefly, the paired-end library was prepared with an insert size of 150 bp using the Illumina paired-end sample preparation kit (Illumina, San Diego, CA) according to the manufacturer's instructions. A HiSeq4000 sequencing platform (Novogene, China) was used, which was generated ∼6.74 million sequence reads from the genomic DNA of Wedge-tailed eagle. The raw datasets were trimmed to pass the quality control based on PHRED score or per base sequence quality score, and the assembly of the mitochondrial genome was conducted according to the established pipeline in CLC Genomics Workbench 9.5.4 under La Trobe University Genomics Platform (Sarker, Das et al. Citation2017; Sarker, Roberts et al. Citation2017). Annotation was performed using Geneious (version 10.2.2), and protein-coding ORFs were further assessed using the CLC Genomics Workbench (version 9.5.4).
The complete mitogenome sequence of A. audax had a circular genome of 17,494 bp, containing 13 protein-coding genes (PCGs), two rRNA genes, 22 tRNA genes, a repeat region and a control region (D-loop) with all protein-coding sequences started with methionine. The contents of A, T, C and G were 30.6.7%, 23.7%, 32.7%, and 13.0%, respectively. AT and GC contents of this complete mitogenome was 54.3% and 45.7%, respectively. The proportion of coding sequences with a total length of 11,007 bp (62.92%), which encodes 3669 amino acids, and all protein-coding genes started with Met. The lengths of 12S and 16S ribosomal RNA were 971 bp and 1545 bp, respectively. The gene arrangement was similar to the complete mitochondrial genomes of other members of the family Accipitridae.
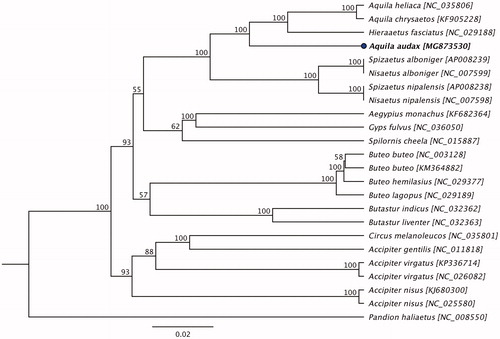
Phylogenetic analysis was performed using complete mitogenome sequence of a A. audax determined in this study with the other mitogenome sequences obtained from the species belonging to the family Accipitridae available in GenBank. The sequences were aligned using the MAFFT L-INS-i algorithm (Katoh et al. Citation2002), and the maximum likelihood (ML) tree with 1000 non-parametric bootstrap resamplings was generated using Geneious (version 10.2.2). As highlighted in , the mitogenome sequence of A. audax was generated a well-separated clade between the two clades; one was dominated by Bonelli's eagle (Jiang et al. Citation2015) and the other was dominated by Hodgson's hawk-eagle and Blyth's hawk-eagle (Asai et al. Citation2006). The highest degree of similarity in nucleotide level (89.01% pairwise) was demonstrated between A. audax and A. heliaca. Therefore, this study concluded that the complete mitogenome of A. audax will be a useful database among the family Accipitridae to study further host-phylogenetic relationship of Accipitridae species, and suggest this may be an implication for the conservation of the species.
Figure 1. Maximum likelihood phylogenetic tree to infer host-phylogeny relationship among Accipitridae family. ML tree was constructed using complete mitogenome sequences of the species belonging to the Accipitridae family. The new complete mitogenome of A. audax is highlighted by bold font.
Disclosure statement
The authors declare no conflicts of interest. The authors alone are responsible for the content and writing of the manuscript. The sequence has been submitted to NCBI under the accession number of MG873530.
References
- Allen D and DeCandido R. 2012. The Eagle Watchers: Observing and Conserving Raptors Around the World. J. Raptor Res. 46:236–238.
- Asai S, Yamamoto Y, Yamagishi S. 2006. Genetic diversity and extent of gene flow in the endangered Japanese population of Hodgson's hawk-eagle, Spizaetus nipalensis. Bird Conserv Int. 16:113–129.
- Das S, Fearnside K, Sarker S, Forwood JK, Raidal SR. 2017. A novel pathogenic aviadenovirus from red-bellied parrots (Poicephalus rufiventris) unveils deep recombination events among avian host lineages. Virology. 502:188–197.
- Doyle JM, Katzner TE, Bloom PH, Ji Y, Wijayawardena BK, DeWoody JA. 2014. The genome sequence of a widespread apex predator, the golden eagle (Aquila chrysaetos). PloS One. 9:e95599.
- Jiang L, Chen J, Wang P, Ren Q, Yuan J, Qian C, Hua X, Guo Z, Zhang L, Yang J, et al. 2015. The mitochondrial genomes of Aquila fasciata and Buteo lagopus (Aves, Accipitriformes): sequence, structure and phylogenetic analyses. PloS One. 10:e0136297.
- Katoh K, Misawa K, Kuma K, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30:3059–3066.
- Knobel J. 2015. The conservation status of the Wedge-tailed Eagle in Australian law and thoughts on the value of early legal intervention in the conservation of a species. De Jure. 48:293–311.
- Sarker S, Das S, Helbig K, Peters A, Raidal SR. 2018. Genome sequence of an Australian strain of canid alphaherpesvirus 1. Aus Veter J. 96:24–27.
- Sarker S, Das S, Lavers JL, Hutton I, Helbig K, Imbery J, Upton C, Raidal SR. 2017. Genomic characterization of two novel pathogenic avipoxviruses isolated from pacific shearwaters (Ardenna spp.). BMC Genomics. 18:298.
- Sarker S, Lloyd C, Forwood J, Raidal SR. 2016. Forensic genetic evidence of beak and feather disease virus infection in a Powerful Owl, Ninox strenua. Emu. 116:71–74.
- Sarker S, Moylan KG, Ghorashi SA, Forwood JK, Peters A, Raidal SR. 2015. Evidence of a deep viral host switch event with beak and feather disease virus infection in rainbow bee-eaters (Merops ornatus). Sci Rep. 5:14511.
- Sarker S, Roberts HK, Tidd N, Ault S, Ladmore G, Peters A, Forwood JK, Helbig K, Raidal SR. 2017. Molecular and microscopic characterization of a novel Eastern grey kangaroopox virus genome directly from a clinical sample. Sci Rep. 7:16472.