
Abstract
The complete chloroplast genome sequence of Homalium cochinchinense was characterized from Illumina pair-end sequencing. The chloroplast genome of H .cochinchinense was 157,116 bp in length, containing a large single-copy region (LSC) of 85,194 bp, a small single-copy region (SSC) of 16,650 bp, and two inverted repeat (IR) regions of 27,636 bp. The overall GC content is 36.6%, while the corresponding values of the LSC, SSC, and IR regions are 64.9%, 69.2%, and 60.3%, respectively. The genome contains 122 complete genes, including 86 protein-coding genes (77 protein-coding gene species), 36 tRNA genes (29 tRNA species) and 8 rRNA genes(4 rRNA species). The Neighbour-joining phylogenetic analysis showed that H. cochinchinense and Flacourtia indica clustered together as sisters to other Salicaceae species, and suggested that this clade may be an ancestral lineage as observed in the phylogenetic tree.
Introduction
Homalium cochinchinense (Lour.) Druce is a member of Salicaceae. Shrubs or small trees, mainly distributed in the southeast of China and Vietnam, which is used in folk medicine for gonorrhaea and as an astringent (Ishikawa et al. Citation2004). In addition to its medicinal uses, this species plays a significant role in the economy and ecology of the areas in which it grows. Nevertheless, wild populations of H. cochinchinense have been destroyed over the course of many years due to overexploitation. Understanding the genetic information of species will help to develop conservation strategies (Zhang et al. Citation2018). In this study, we characterized the whole chloroplast genome of H. cochinchinense and learned more about genetic information of this species, which can contribute to the conservation, and provide useful help for population genetics studies of H. cochinchinense.
The fresh leaves of H.cochinchinense were collected from Guangzhou (Guangdong, China; 23°04′N, 113°20′E), And voucher specimen is deposited at the herbarium of Research Institute of Forestry. Total genomic DNA was isolated from silica-gel-dried leaves using the modified CTAB method (Doyle JJ and Doyle JL Citation1987). Paired-end sequencing libraries with an insert size of 500 bp were constructed according to the Illumina library preparation protocol. Sequencing was carried out on the Illumina HiSeq 2000 platform (Illumina, San Diego, CA). We assembled the chloroplast genome using MITObim 1.8 (University of Oslo, Oslo, Norway; Kaiseraugst, Switzerland) (Hahn et al. Citation2013), with Flacourtia indica (GenBank: MG262341) as the reference. DOGMA software (Wyman et al. Citation2004) was used for annotation of the complete chloroplast genome, and the annotation was corrected with Geneious 8.0.2 (Kearse et al. Citation2012) and Sequin 15.50 (http://www.ncbi.nlm.nih.gov/Sequin/).
The complete chloroplast genome of H. cochinchinense (GenBank accession number MK123970) is 157,116 bp in length, containing a large single-copy region (LSC) of 85,194 bp, a small single-copy region (SSC) of 16,650 bp, and two inverted repeat (IR) regions of 27,636 bp. The overall GC content is 36.6%, while the corresponding values of the LSC, SSC, and IR regions are 64.9%, 69.2%, and 60.3%, respectively. The chloroplast genome contains 122 complete genes, including 86 protein-coding genes (77 protein-coding gene species), 36 tRNA genes (29 tRNA species) and 8 rRNA genes (4 rRNA species). Most of the genes occur as a single copy, except for 20 gene species occur in double copies, including nine protein-coding genes (rps7, rps12, rpl2, rpl23, rps19, ndhB, ycf1,ycf2, and ycf15), seven tRNAs (trnA-UGC, trnI-CAU, trnI-GAU, trnL-CAA, trnN-GUU, trnR-ACG, and trnV-GAC), and four rRNAs (4.5S, 5S, 16S, and 23S rRNA).
Phylogenetic analysis was performed using complete chloroplast genome of H. cochinchinense, 10 related species of Salicaceae, and 2 species of Passifloraceae (a family closely related to Salicaceae), with Hevea brasiliensis (Euphorbiaceae) as the outgroup. Neighbour-joining tree (with 1000 bootstrap replicates) was generated using MEGA 7.0 (Kumar et al. Citation2016) from alignments created by the MAFFT (Katoh and Standley Citation2013). The results showed that H. cochinchinense and F. indica clustered together as sisters to other Salicaceae species, and suggested that this clade may be an ancestral lineage as observed in the phylogenetic tree ().
Figure 1. Neighbour-joining (NJ) analysis of H. cochinchinense and other related species based on the complete chloroplast genome sequence. The gene’s accession number is listed in the figure.
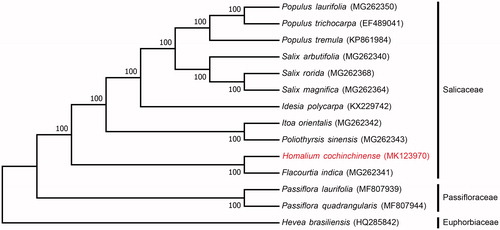
In conclusion, the complete chloroplast genome from this study not only sheds light on conservation for H. cochinchinense, but facilitates the phylogenetic studies of the Salicaceae family.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19:11–15.
- Hahn C, Bachmann L, Chevreux B. 2013. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads-a baiting and iterative mapping approach. Nucleic Acids Res. 41:e129.
- Ishikawa T, Nishigaya K, Takami K, Uchikoshi H, Chen IS, Tsai IL. 2004. Isolation of salicin derivatives from Homalium cochinchinensis and their antiviral activities. J Nat Prod. 67:659–663.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30:772–780.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28:1647–1649.
- Kumar S, Stecher G, Tamura K. 2016. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 33:1870–1874.
- Wyman SK, Jansen RK, Boore JL. 2004. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 20:3252–3255.
- Zhang L, Guo XY, Wang ZF, Wang MC, Hu QJ. 2018. Characterization of the complete chloroplast genome of Musella lasiocarpa. Mitochondrial DNA Part B. 3:728–729.