
Abstract
Sequencing the Sophora flavescens mitochondrial genome is important for elucidating the evolutionary characteristics of traditional Chinese medical plants. In this study, the complete mitochondrial (mt) genome of S. flavescens was sequenced through Illumina sequencing method, and the mitochondrial genome map was constructed after de novo assembly and annotation. The results showed that the 494,369 bp S. flavescens mitochondrial genome has a total of 78 genes, including 53 protein-coding genes, three rRNA genes, and 23 tRNA genes. The overall GC content of this mitogenome is 44.9%. By phylogenetic analysis using maximum likelihood method, S. flavescens showed the closest relationship with Vigna radiata in the clade of Papilionoideae.
The shrubby sophora (Sophora flavescens) is a species of plant in the genus Sophora, which belongs to the Fabaceae family. It is a traditional Chinese medicine that has been used for anti-tumor, viral hepatitis, enteritis, viral myocarditis, arrhythmia, and skin diseases. Here, we assembled and analyzed the mitochondrial genome of S. flavescens based on the next-generation sequencing method.
Plant materials of S. flavescens Ait. sequenced in this study were acquired from medical plants garden in Guiyang University of Traditional Chinese Medicine (26°57'N, 106°72'W). Both the materials and its total genomic DNA and were stored in the Key Laboratory of Miao Medicine, Guiyang University of Traditional Chinese Medicine. Sequencing was done on an Illumina HiSeq2500 platform. The clean reads were assembled using SPAdes 3.11.1 (Bankevich et al. Citation2012) with default settings.
We used other mitochondrial genomes from higher plants as seeds to collect the mitochondrial contigs as much as possible. To fill the gap, Price (Ruby et al. Citation2013) and MITObim v1.8 (Hahn et al. Citation2013) were applied and Bandage (Wick et al. 2015) was used to construct the circular assembly path guided by these positions of long scaffolds. The complete sequence was primarily annotated using Plann (Huang and Quentin 2015) and Exonerate (Slater Guy St. C. and Birney Ewan Citation2005) combined with manual correction. All tRNAs were confirmed using the tRNAscan-SE search server (Lowe and Sean 1997). Other protein-coding genes were verified using BLAST search on the NCBI website, and manual correction for start and stop codons was conducted. This complete mitochondrial genome sequence was submitted to GenBank under the accession numbers of MH922916.
Mitochondrial genome of S. flavescens Ait. is a typical circular structure of 494,369 bp with a GC content of 44.9%. This mitochondrial genome contains a total of 78 genes, including 53 protein-coding genes, three rRNA genes (rrn5, rrn18, and rrn26), 17 complete native mitochondrial tRNA genes, and six chloroplast-derived tRNA genes.
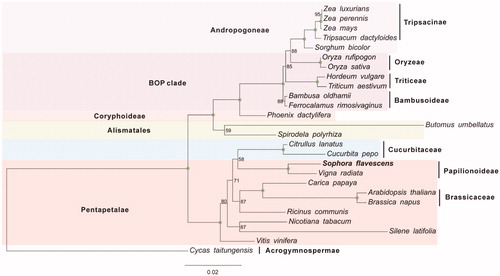
Another 25 published complete mitochondrial sequences of Spermatophyta were downloaded from the NCBI database. Eight shared protein-coding genes were selected for phylogenetic analysis. These nucleotide sequences were separately aligned using PRANK (Löytynoja Ari and Nick Goldman Citation2010). After trimming using Gblocks (Dereeper et al. Citation2008), the independent alignments were subsequently concatenated. ModelFinder (Kalyaanamoorthy et al. Citation2017) was used to identify the most appropriate substitution model. Phylogenetic analyses of the concatenated sequences were performed using the maximum likelihood (ML) method implemented in IQ-TREE version 1.6.6 (Nguyen et al. Citation2014) according to the optimal substitution models under the rapid bootstrap algorithm (1000 replicates). To test support for the branch points of each gene trees, nonparametric branch support tests based on the Shimodaira–Hasegawa-like approximate likelihood ratio test (SH-like aLRT) procedure were also performed in the same run. As shown in , the phylogenetic positions of these 26 mt genomes were successfully resolved with full bootstrap supports across almost all nodes. S. flavescens Ait., belonging to the Sophoreae, exhibited the closest relationship with Vigna radiata in the clade of Papilionoideae.
Figure 1. Phylogenetic tree yielded by ML analysis of 26 higher plant mt genomes. ML consensus tree is shown with bootstrap supports indicated by numbers besides branches.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19:455–477.
- Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, Dufayard J-F, Guindon S, Lefort V, Lescot M, et al. 2008. Phylogeny. fr: robust phylogenetic analysis for the non-specialist. Nucl Acids Res. 36:W465–W469.
- Hahn C, Lutz B, Bastien C. 2013. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads-a baiting and iterative mapping approach. Nucl Acids Res. 41:e129–e129.
- Huang DI, Quentin CBC. 2015. Plann: a command-line application for annotating plastome sequences. Appl Plant Sci. 3:1500026.
- Kalyaanamoorthy S, Minh BQ, Wong TKF, Haeseler A, von Jermiin LS. 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 14:587.
- Lowe TM, Sean RE. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucl Acids Res. 25:955–964.
- Löytynoja A, Nick G. 2010. webPRANK: a phylogeny-aware multiple sequence aligner with interactive alignment browser. BMC Bioinform. 11:1579.
- Nguyen L-T, Schmidt HA, Haeseler A, von Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32:268–274.
- Ruby JG, Priya B, Joseph L. DRisi. 2013. PRICE: software for the targeted assembly of components of (Meta) genomic sequence data. G3. 3:865–880.
- Slater GSC, Birney E. 2005. Automated generation of heuristics for biological sequence comparison. BMC Bioinform. 6:31.
- Wick RR, Schultz MB, Zobel J, Holt KE. 2015. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics. 31:3350–3352.