
Abstract
Lepisorus waltonii (Ching) S. L. Yu is a typical alpine fern species in the Qinghai-Tibet Plateau and Hengduan Mountain Regions. Here, we described the complete chloroplast genome of L. waltonii using Illumina paired-end sequencing. The chloroplast genome length is 157,148 bp, containing two inverted repeats (IR, 27,110 bp each) regions, separated by a large single-copy (LSC, 81,243 bp), and a small single-copy (SSC, 21,685 bp) regions, respectively. The genome contains 118 functional genes, including 85 protein-coding genes, 29 tRNA genes, and four rRNA genes. The total GC content of the chloroplast genome is 41.8%. ML phylogenetic analysis revealed that L. waltonii was closest related to Lepisorus clathratus.
Lepisorus waltonii (Ching) S.L. Yu is a perennial, epilithic fern mainly distributed in the Qinghai-Tibet Plateau, growing in crevices of natural rocks at an elevation of 3400–5000 m (Ching and Wu Citation1980; Wang et al. Citation2011). Its unusual morphology and considerable variation lead to the controversy of the taxonomy and status of this species (Qi et al. Citation2013). Recently, the phylogenetic results supported L. waltonii as a member of L. clathratus complex (Wang et al. Citation2010). While the cross-amplification of microsatellite markers suggested, it is a recent divergence from L. clathratus (Zhao et al. Citation2016). Here, we describe the chloroplast genome of L. waltonii and determine its phylogenetic location, contributing to the taxonomy and status of L. waltonii.
Leaf material of L. waltonii was collected in Lhasa, Tibet (29.71303°N, 91.09942°E). The voucher specimen (C.F. Zhao NR6) has been deposited in the Herbarium of the Institute of Botany, Chinese Academy of Sciences. Total genomic DNA was isolated using Tiangen Plant Genomic DNA Kit (Tiangen Biotech Co., Beijing, China). The NEBNext DNA Library Prep Kit (New England Biolabs, Ipswich, MA) was used for library construction. Paired-end reads of 2 × 150 bp were generated with Illumina HiSeq PE150 (Illumina, San Diego, CA). The chloroplast genome data were extracted using L. clathratus (KY419704) as a reference and assembled de novo with Geneious (Kearse et al. Citation2012). The initial annotation was performed using DOGMA (Wyman et al. Citation2004). After initial annotation, the putative starts, stops, and intron positions were determined by comparison with homologous genes in L. clathratus chloroplast genomes (Wei et al. Citation2017). The tRNA genes were annotated using DOGMA and tRNAscan-SE (Schattner et al. Citation2005). The circular chloroplast genome map was drawn with OGDraw (Lohse et al. Citation2013).
The size of the L. waltonii chloroplast genome (GenBank accession MK287776) is 157,148 bp, including a pair of inverted repeat (IR) regions of 27,110 bp each, separated by a large single-copy (LSC) region of 81,243 bp and a small single-copy (SSC) region of 21,685 bp. In total, 85 protein-coding genes (PCGs), 29 tRNA genes and four rRNA genes are successfully annotated. Among them, five PCGs, five tRNA genes, and four rRNA genes are duplicated in the IR regions. Among these genes, 11genes (clpP, rpl2, rpl16, petD, trnI-GAU, trnA-UGC, trnL-UAA, rps3, rpl22, rps19, trnG-GCC) contain one intron, one gene (ndhA) contains two introns. The GC content of the whole chloroplast genome is 41.8%, greater than that of the LSC (40.4%) and SSC (37.9%) regions.
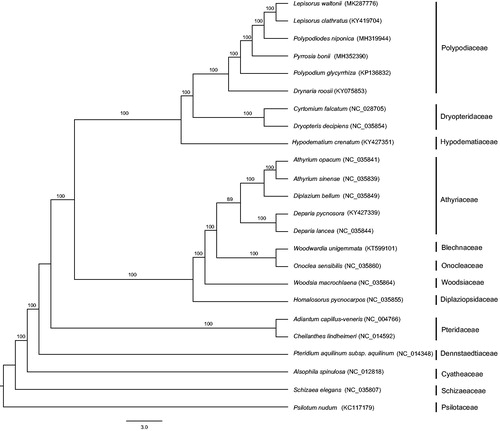
Phylogenetic analysis based on 83 protein-coding genes was performed (Psilotum nudum as outgroup). The sequences were aligned using MAFFT (Katoh et al. Citation2002), and phylogenetic tree was inferred based on the maximum likelihood (ML) analysis using RAxML with 1000 bootstrap replicates (Stamatakis Citation2014). ML tree indicates that L. waltonii is most closely related to L. clathratus with strong support (). The sequence variation rate between L. waltonii and L. clathratus is 0.4679%. These results and the distinct morphological characters of L. waltonii support it should be treated as an independent species.
Figure 1. Phylogenetic tree based on 83 protein-coding genes using the ML method. Bootstrap values are shown above the nodes, with 1000 bootstrap replicates.
Acknowledgments
We thank Hong-Rui Zhang and Jong-Soo Kang for the assistance in data analyses.
Disclosure statement
The authors report no conflicts of interest and are solely responsible for the content and writing of this paper.
Additional information
Funding
References
- Ching R-C, Wu S-K. 1980. Platygyria Ching et S. K. Wu, an unique new genus of the Polypodiaceae from China. Acta Bot Yunnan. 2:67–74.
- Katoh K, Misawa K, Kuma K, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30:3059–3066.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28:1647–1649.
- Lohse M, Drechsel O, Kahlau S, Bock R. 2013. OrganellarGenomeDRAW—a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41:575–581.
- Qi X-P, Zhang X-C, Lin Y-X, Gilbert MG, Hovenkamp PH. 2013. Lepisorus (J. Smith) Ching. In: Wu CY, Raven P, Hong D-Y, editors. Flora of China, Vol. 2–3. Beijing: Science Press; p. 808–824. Missouri Botanical Garden Press, St. Louis.
- Schattner P, Brooks AN, Lowe TM. 2005. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33:W686–W689.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30:1312–1313.
- Wang L, Qi XP, Xiang QP, Heinrichs J, Schneider H, Zhang XC. 2010. Phylogeny of the paleotropical fern genus Lepisorus (Polypodiaceae, Polypodiopsida) inferred from four chloroplast DNA regions. Mol Phyl Evol. 54:211–225.
- Wang L, Wu ZQ, Bystriakova N, Ansell SW, Xiang QP, Heinrichs J, Schneider H, Zhang XC. 2011. Phylogeography of the Sino-Himalayan fern Lepisorus clathratus on “the roof of the world”. PLoS One. 6:e25896.
- Wei R, Yan YH, Harris AJ, Kang JS, Shen H, Xiang QP, Zhang XC. 2017. Plastid phylogenomics resolve deep relationships among eupolypod II ferns with rapid radiation and rate heterogeneity. Genome Biol Evol. 9:1646–1657.
- Wyman SK, Jansen RK, Boore JL. 2004. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 20:3252–3255.
- Zhao CF, Kwak M, Xiang QP. 2016. Isolation and characterization of microsatellite markers in the Lepisorus clathratus complex (Polypodiaceae). Appl Plant Sci. 4:1600069.