
Abstract
Prunus matuurai is an endemic flowering cherry on Taiwan Island, China. Here, we determined the first complete chloroplast genome of P. matuurai using genome skimming approach. The cp genome was 157,928 bp long, with a large single-copy region (LSC) of 85,904 bp and a small single-copy region (SSC) of 19,104 bp separated by a pair of inverted repeats (IRs) of 26,460 bp. It encodes 129 genes, including 84 protein-coding genes, 37 tRNA genes, and 8 ribosomal RNA genes. The phylogenetic analysis indicated that P. matuurai is closely related to P. speciose.
Prunus matuurai Sasaki is an indigenously wild flowering cherry species of Taiwan Island. Its natural distribution is restricted to its type locality, Mt. Taipingshan at alt ca. 2000m (Li Citation1963; Hsieh Citation1993) and a sparse distribution on neighboring mountains in Hualian county (according to specimens deposited in Herbarium, Biodiversity Research Center, Academia Sinica, Taipei (HAST) and Herbarium of Taiwan Forestry Research Institute (TAIF)). According to Sasaki, P. matuurai is morphologically close to P. transarisanensis Hayata, a species also endemic to Taiwan island (Li Citation1963; Hsieh Citation1993), but the relationship of P. matuurai with other flowering cherries has not been established, the taxonomic status in Prunus is still uncertain (Li and Bartholomew Citation2003). Therefore, we sequenced the whole chloroplast genome of P. matuurai to elucidate its phylogenetic relationship with other flowering cherries.
Total genomic DNA was extracted from silica-dried leaves collected from Mt. Taipingshan in the northeast of Taiwan Island using a modified CTAB method (Doyle and Doyle Citation1987). The voucher specimen (Sun1708035) was collected and deposited in the Herbarium of Zhejiang Academy of Forestry (HZJAF). DNA libraries preparation and pair-end 125 bp read length sequencing were performed on the Illumina HiSeq 2500 platform. About 6.6 Gb of raw data were trimmed and assembled into contigs using CLC Genomics Workbench 8. Then, all the contigs were mapped to the reference cp genome of Prunus takesimensis (MG754959; Cho et al. Citation2018) using BLAST (NCBI BLAST v2.2.31) search and the draft cp genome of P. matuurai was constructed by connecting overlapping terminal sequences in Geneious R11 software (Biomatters Ltd., Auckland, New Zealand). Gene annotation was performed via the online program Dual Organellar Genome Annotator (DOGMA; Wyman et al. Citation2004).
The complete cp genome of P. matuurai (GenBank accession MK239267) was 157,928 bp long, consisting of a pair of inverted repeat regions (IRs with 26,460 bp) divided by two single-copy regions (LSC with 85,904 bp; SSC with 19,104 bp). The overall GC content of the total length, LSC, SSC, and IR regions were 36.7%, 34.6%, 30.2%, and 42.5%, respectively. The genome contained a total of 129 genes, including 84 protein-coding genes, 37 tRNA genes, and 8 rRNA genes.
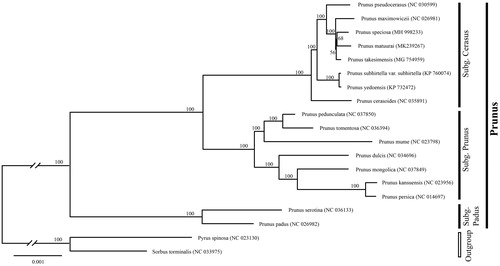
To determine the phylogenetic position of newly sequenced P. matuurai, phylogenetic analysis was conducted along with 17 representative Prunus L. species and two outgroup taxa. We reconstructed a phylogeny employing the GTR + G model and 1000 bootstrap replicates under the maximum-likelihood (ML) inference in RAxML-HPC v.8.2.10 on the CIPRES cluster (Miller et al. Citation2010). The ML tree () was consistent with the most recent phylogenetic study on Prunus (Shi et al. Citation2013; Chin et al. Citation2014). P. matuurai belongs to subg. Cerasus as expected and exhibited the closest relationship with Prunus speciose (Koidz.) Nakai.
Figure 1. Phylogenetic tree reconstruction of 17 taxa of Prunus and two outgroups using ML method. Relative branch lengths are indicated. Numbers near the nodes represent ML bootstrap value.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Chin SW, Shaw J, Haberle R, Wen J, Potter D. 2014. Diversification of almonds, peaches, plums and cherries – molecular systematics and biogeographic history of Prunus (Rosaceae). Molec Phylogen Evol. 76:34–48.
- Cho MS, Yang JY, Kim SC. 2018. Complete chloroplast genome of Ulleung Island endemic flowering cherry, Prunus takesimensis (Rosaceae), in Korea. Mitochondrial DNA B. 3:274–275.
- Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19:11–15.
- Hsieh CF. 1993. Prunus. In: Huang TC, editor. Flora of Taiwan, 2nd Edition. Taipei: National Taiwan University; p. 96–104.
- Li CL, Bartholomew B. 2003. Cerasus. In: Wu ZY, Raven PH, editors. Flora of China, Vol. 9. Beijing: Science Press; St. Louis: Missouri Botanical Garden Press; p. 404–420.
- Li HL. 1963. Woody flora of Taiwan. Narberth (Pennsylvania): Livingston Pub. Co.
- Miller MA, Pfeiffer W, Schwartz T. 2010. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. Gateway Comput Environ Workshop. 14:1–8.
- Shi S, Li J, Sun J, Yu J, Zhou S. 2013. Phylogeny and classification of Prunus sensu lato (Rosaceae). J Integrat Plant Biol. 55:1069–1079.
- Wyman SK, Jansen RK, Boore JL. 2004. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 20:3252–3255.