
Abstract
This study was the first report about the complete chloroplast genome of Aster hersileoides (Asteraceae, Astereae). The circular whole cp genome of A. hersileoides was 152,345 bp in length, contained a large single copy (LSC) region of 84,124 bp and a small single copy (SSC) region of 18,243 bp. These two regions were separated by a pair of inverted repeat regions (IRa and IRb), each of them 24,989 bp in length. A total of 134 functional genes were encoded, consisted of 89 protein-coding genes, 37 tRNA genes, and 8 rRNA genes. The overall GC content of the chloroplast genome sequence was 37.3% and the GC contents of the LSC, SSC, and IR regions were 32.6, 31.2, and 43.0%, respectively. The phylogenetic hypotheses obtained based on the analyses of 20 cp genomes places A. hersileoides within the tribe Astereae of Asteraceae.
The genus Aster (Asteraceae) and its related genera represent one of the most species-rich lineages of tribe Astereae, about 152 species are known, of which more than 80% are distributed in China. The shrub Aster hersileoides is a Chinese endemic species that has a restricted distribution in limestone regions of Sichuan province (Chen et al. Citation2011), the recent phylogenetic analysis demonstrated that this species should be removed from Aster and deserves generic status (Li et al. Citation2012). A good knowledge of genomic information of this species would contribute to the formulation of protection strategy and the study of genome diversity and species diversity. In this study, we assembled and characterized its complete chloroplast genome (Gen-Bank accession no. MK290823) from Illumina sequencing data.
Fresh leaves of A. hersileoides were collected from Lixian, Sichuan Province, China and deposited in Herbarium, Sichuan Normal University, SCNU (specimen no.: Z.X. Fu 3049). High-quality total genomic DNA was extracted from ca.6 cm2 sections of the silica-dried leaf using improved Tiangen Plant Genomic DNA Kits (DP305) based on manual (TIANGEN Biotech (Beijing) Co., Ltd.), add the 4 μl RNAseA and 20 μl Proteinase K after incubated (65 °C). Total DNA was directly constructed short-insert of 150 bp in length libraries and sequenced on the Illumina Genome Analyzer (Hiseq 2000, Illumina, San Diego, CA, USA) based on the manufacturer’s protocol by ORI-GENE, Beijing, China. Generally, more than 6 Gb of data was obtained for the complete cp genome of A. hersileoides, De novo assembled in CLC Genomic Workbench v11 (CLC Bio, Aarhus, Denmark) and consensus sequence in Geneious R11.1.5 (Biomatters Ltd., Auckland, New Zealand) with referenced chloroplast genome sequence of Conyza bonariensis H.B.K (Accession: KX792499). The chloroplast genome was annotated using a web-based annotation program GeSeq (https://chlorobox.mpimp-golm.mpg.de/geseq.html) and editing by manual and imagining with OGDraw v1.2 (Lohse et al. Citation2013).
The complete chloroplast genome of A. hersileoides (GenBank Accession No. MK290823) was 152,345 bp in length and a typical circular structure comprising a pair of inverted repeat (IR) of 24,989 bp divided by a large single copy (LSC) region of 84,124 bp and a small single copy (SSC) region of 18,243 bp (). The general G + C content was 37.3% in the whole sequence, while corresponding values of 32.6, 31.2, and 43.0% in the LSC, SSC, and IR regions. The whole genome contained 134 genes, including 89 protein-coding genes, 8 ribosomal RNA genes, and 37 tRNA genes. Nevertheless, 114 unique genes, 20 genes duplicated in the IRs. In addition, among the annotated chloroplast genomic sequence, 15 genes possessed only single intron, two genes (ycf3 and clpP) possessed two introns.
Figure 1. The best maximum likelihood (ML) phylogram inferred from 20 chloroplast genomes in Asteraceae (bootstrap value are indicated on the branches).
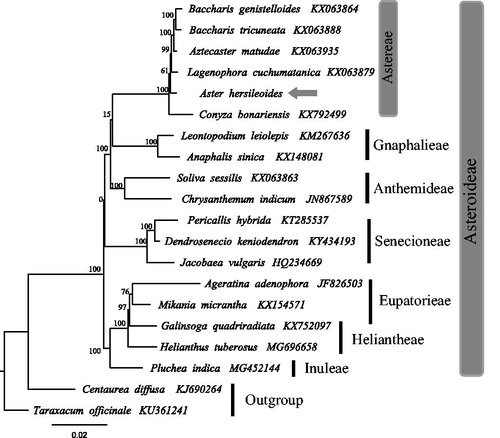
To construct the phylogenetic tree, all of the cp genome sequences were aligned in MAFFT (Katoh and Standley Citation2013). A maximum likelihood analysis based on the GTRGAMMA model was performed with RaxML v7.2.8 on the CIPRES (Stamatakis et al. Citation2008; Miller et al. Citation2010) using 1000 bootstrap replicates with Centaurea diffusa Lam. and Taraxacum officinale (Asteraceae) as outgroup. Astereae species form a clade with a high bootstrap value (BP = 100) and Aster hersileoides formed a basally branching within the clade (Lagenophora cuchumatanica, Baccharis genistelloides, Baccharis tricuneata).
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Additional information
Funding
References
- Chen YL, Brouillet L, Semple JC. 2011. Aster L. In: Wu CY, Raven PH, and Hong DY, editors. Flora of China. Vol. 20–21. Beijing and Missouri: Science Press; St. Louis: Botanical Garden Press. p. 574–632.
- Li WP, Yang FS, Jivkova T, Yin GS. 2012. Phylogenetic relationships and generic delimitation of Eurasian Aster (Asteraceae: Astereae) inferred from ITS, ETS and trnL-F sequence data. Ann Bot. 109:1341–1357.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30:772–780.
- Lohse M, Drechsel O, Kahlau S, Bock R. 2013. Organellar Genome DRAW—a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucl Acids Res. 41:575–581.
- Miller MA, Pfeiffer W, Schwartz T. 2010. Creating the CIPRES science gateway for inference of large phylogenetic trees. Proceedings of the gateway computing environments workshop (GCE). New Orleans (LA):IEEE. p. 1–8.
- Stamatakis A, Hoover P, Rougemont J. 2008. A rapid bootstrap algorithm for the RAxML Web servers. Syst Biol. 57:758–771.