
Abstract
The whole chloroplast (cp) genome sequence of Carrierea calycina has been characterized using Illumina pair-end sequencing. The complete cp genome was 158,085 bp in length, containing a large single copy region (LSC) of 84,663 bp, and a small single copy region (SSC) of 17,570 bp, which were separated by a pair of 27,926 bp inverted repeat regions (IRs). The genome contained 129 genes, including 85 protein-coding genes, 36 tRNA genes, and eight rRNA genes. The overall GC content of C. calycina cp genome is 36.8%, whereas the corresponding values of the LSC, SSC, and IR regions are 34.8%, 30.3%, and 41.8%, respectively. Further, phylogenetic analysis suggested that the location of C. calycina is out of the Idesia polycarpa and it is an outgroup to the genera of Salix and Populus.
Carrierea calycina, which is native to China, is a species of tree in the willow family. The wood of C. calycina can be used to make furniture and other utensils. The industrial oil can be squeezed from the seeds. At present, there are few studies on C. calycina, understanding the genomic characteristics of it is important for the evolutionary analysis of Salicaceae. In this study, we assembled and characterized the complete chloroplast genome sequence of C. calycina based on the Illumina pair-end sequencing data. The voucher specimen was collected at Jiangkou Xian, Daiyenpeng along the Kaitu River on the SW side of the Fanjing Shan mountain range (108。E,27。N) and stored at Arnold Arboretum of Harvard University.
The total genomic DNA was extracted from fresh leaves using a modified CTAB method (Doyle and Doyle Citation1987). The filtered reads were assembled using the program NOVOPlasty (Dierckxsens et al. Citation2017) for C. calycina using the sequenced plastome of its close relative Itoa orientalis (GenBank accession no. MG262342) as the reference. Next, the assembled plastome was annotated using Plann (Huang and Cronk Citation2015), and the annotation was corrected using Geneious (Kearse et al. Citation2012). In addition, the physical map of the plastome was generated using OGDRAW (Lohse et al. Citation2013). The complete plastome together with gene annotations was submitted to GenBank under the accession number of MK263737. To further investigate its phylogenetic placement, a neighbor-joining tree (Saitou and Nei Citation1987) was constructed based on complete plastome sequences of 15 seed plants. Here, DNA sequences from these 15 plastomes were first aligned using MAFFT (Katoh and Standley Citation2013), and phylogenetic tree was then constructed using MEGA7 (Kumar et al. Citation2016) with 1000 bootstrap replicates.
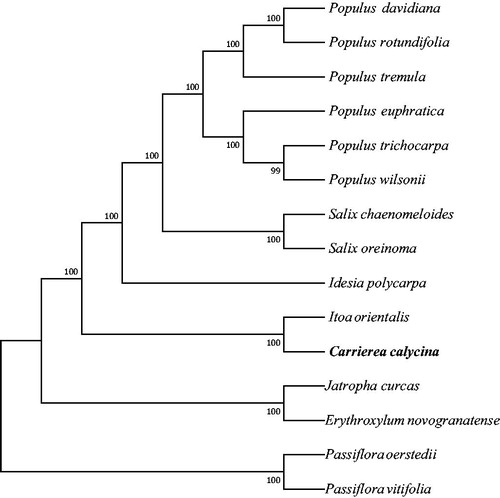
Our assembled plastome of C. calycina is 158,085 base pairs (bp) in size, containing a large single-copy (LSC) region of 84,663 bp, a small single-copy (SSC) region of 17,570 bp, and two inverted repeat (IR) regions of 27,926 bp. In addition, the plastome possesses total 129 genes, including 85 protein-coding genes, eight rRNA genes, and 36 tRNA genes. Most of these genes are in a single copy, however, four rRNA genes (i.e., 4.5S, 5S, 16S, and 23S rRNA), seven protein-coding genes (i.e., ndhB, rpl2, rpl23, rps12, rps19, rps7, and ycf2), and seven tRNA genes (i.e., trnA-UGC, trnI-CAU, trnI-GAU, trnL-CAA, trnN-GUU, trnR-ACG, and trnV-GAC) occur in double copies. The overall GC-content of the whole plastome is 36.8%, whereas the corresponding values of the LSC, SSC, and IR regions are 34.8%, 30.3%, and 41.8%, respectively. Furthermore, the location of C. calycina is out of the Idesia polycarpa and it is an outgroup to the genera of Salix and Populus with a over 90% bootstrap support (). The location of some species is consistent with previous study (Zhang et al. Citation2018).
Figure 1. Phylogenetic relationships of 15 seed plants based on plastome sequences. Bootstrap percentages are indicated for each branch. GenBank accession numbers: Erythroxylum novogranatense (KX256287), Passiflora vitifolia (MF807947.1), Passiflora oerstedii (MF807942.1), Idesia polycarpa (KX229742), Itoa orientalis (MG262342), Jatropha curcas (FJ695500), Populus tremula (KP861984.1), Populus davidiana (KX306825.1), Populus rotundifolia (KX425853.1), Populus euphratica (KJ624919), Populus trichocarpa (EF489041), Populus wilsonii (MG262359), Salix chaenomeloides (MG262362), and Salix oreinoma (MF189168.1).
The complete plastome can provide valuable information to develop highly variable DNA markers for population genetic survey, species delimitation, and other ecological and evolutionary studies. In addition, our study will benefit the study of the relationships between Salicaceae species.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Additional information
Funding
References
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45:e18.
- Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19:11–15.
- Huang DI, Cronk QCB. 2015. Plann: a command-line application for annotating plastome sequences. Appl Plant Sci. 3:1500026.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30:772–780.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28:1647–1649.
- Kumar S, Stecher G, Tamura K. 2016. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger Datasets. Mol Biol Evol. 33:1870–1874.
- Lohse M, Drechsel O, Kahlau S, Bock R. 2013. Organellar Genome DRAW-a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41:W575–W581.
- Saitou N, Nei M. 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 4:406–425.
- Zhang L, Xi Z, Wang M, Guo X, Ma T. 2018. Plastome phylogeny and lineage diversification of Salicaceae with focus on poplars and willows. Ecol Evol. 8:7817–7823.