
Abstract
The first complete chloroplast genome of Costus viridis (Costaceae) was reported in the current study. The C. viridis genome was 168,966 bp in length and comprised a pair of inverted repeat (IR) regions of 29,166 bp each, a large single-copy (LSC) region of 92,189 bp, and a small single-copy (SSC) region of 18,445 bp. It encoded 133 genes, including 87 protein-coding genes (79 PCG species), 38 tRNA genes (28 tRNA species), and eight rRNA genes (four rRNA species). The overall AT content was 63.75%. Phylogenetic analysis showed that C. viridis was closely related to species Costus osae within the genus Costus in family Costaceae.
Costus viridis, endemic to southwest China, is one species of the genus Costus (Costaceae), which distributes in Yunnan province and grows in moist places in forests (1000 m) (Wu and Larsen Citation2000). Its stem length is 2–3 m; leaf sheaths are green; inflorescences are terminal and broadly ovoid; bracts are imbricate, green, ovate or broadly, leathery, margin membranous, apex brown and sharply pointed; lobes are golden, rigid, and glabrescent at apex (Wu and Larsen Citation2000). This species owns high ornamental value and can be used as courtyard ornamental and landscape engineering plants (Wu et al. Citation2016). However, this species has been endangered for ever-increasing habitat degradation or loss and thus has been categorized into the red list of Chinese plants (http://www.chinaplantredlist.org/search.php). So far, genetic information of C. viridis is largely lacking; therefore, its complete chloroplast genome sequence is reported here. It would be useful for the research on the phylogenetic relationships of C. viridis and species identification within Costaceae.
Costus viridis was collected from Wanding Zhen, Yunnan province and stored at the resource garden of environmental horticulture research institute, Guangdong academy of agricultural sciences, Guangzhou, China. Total chloroplast DNA was extracted from about 100 g of fresh leaves of C. viridis using the sucrose gradient centrifugation method (Li et al. Citation2012). Library construction was using Illumina (Illumina, CA, USA) and PacBio (Novogene, Beijing, China) sequencing, respectively. In total, 66.4 M clean data of 150 bp paired-end reads and 0.92 M clean data of 8–10 kb subreads were generated. The Illumina clean reads were assembled using SOAPdenova (version 2.04) with default parameters into principal contigs (Luo et al. Citation2012), and all contigs were sorted and joined into a single draft sequence using the software Geneious version 11.0.4 (Kearse et al. Citation2012). Next, BLASR software was used to compare the PacBio clean data with the single draft sequence and to extract the correction and error correction (Chaisson and Tesler Citation2012). Next, the corrected PacBio clean data were assembled using Celera Assembler (version 8.0) with default parameters, thus generating scaffolds (Denisov et al. Citation2008). Next, the assembled scaffolds were mapped back to the Illumina clean reads using GapCloser (version 1.12) for gap closing (Luo et al. Citation2012). Finally, the redundant fragments sequences were removed, thus generating the final assembled chloroplast genomic sequence. The assembled chloroplast genome was annotated using the online tool DOGMA (Dual Organellar Genome Annotator) (Wyman et al. Citation2004) with default parameters and checked manually. tRNA genes were identified using tRNAscanSE with default settings (Lowe and Chan Citation2016). The circular map of the chloroplast genome of C. viridis was drawn using OGDRAWv1.2 (Lohse et al. Citation2013). The assembled and annotated chloroplast genome sequence has been submitted to GenBank (accession no. MK262733).
The complete chloroplast genome of C. viridis was 168,966 bp in length and comprised a pair of inverted repeat (IR) regions of 29,166 bp each, a large single-copy (LSC) region of 92,189 bp and a small single-copy (SSC) region of 18,445 bp. It was predicted to contain a total of 133 genes, including 38 tRNA (28 tRNA species), eight rRNA (four rRNA species), and 87 protein-coding genes (79 PCG species). Twenty gene species occurred in double copies, including eight PCG species (ndhB, rpl2, rpl22, rpl23, rps7, rps12, rps19, and ycf2), eight tRNA species (trnH-GUG, trnI-CAU, trnL-CAA, trnV-GAC, trnI-GAU, trnA-UGC, trnR-ACG, and trnN-GUU), and all four rRNA species (rrn4.5, rrn5, rrn16, and rrn23). All these 20 gene species were located in the IR regions. The ycf1 gene crossed the bounder of SSC and IRb region, whereas the rps12 gene was located its first exon in the LSC region and other two exons in the IRs regions. In addition, 10 PCG genes (atpF, ndhA, ndhB, rpoC1, petB, petD, rpl2, rpl16, rps12, and rps16) and 6 tRNA genes (trnK-UUU, trnG-GCC, trnL-UAA, trnV-UAC, trnI-GAC, and trnA-UAC) had a single intron, whereas two other genes (ycf3 and clpP) possessed two introns. The nucleotide composition was asymmetric (31.67% A, 18.35% C, 17.91% G, 32.08% T) with an overall AT content of 63.75%. The AT contents of the LSC, SSC, and IR regions were 65.94%, 70.4%, and 58.18%, respectively.
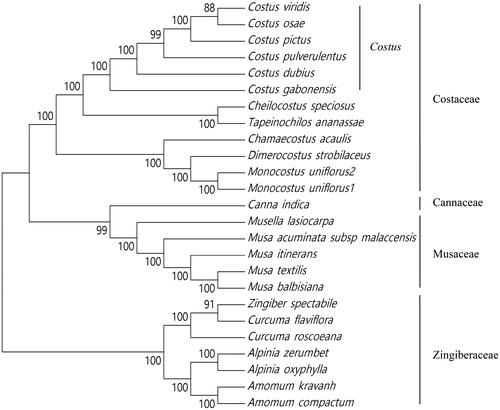
To obtain its phylogenetic position within the order Zingiberales, the chloroplast genome of C. viridis was aligned with those of other 24 species from four families, Costaceae, Zingiberaceae, Cannaceae, and Musaceae, using MAFFT (Katoh and Standley Citation2013). A maximum likelihood phylogenetic tree () was constructed using the 25 chloroplast genome sequences alignment result with MEGA7 (Kumar et al. Citation2016). The phylogenetic tree showed that C. viridis was closely related to species Costus osae within the genus Costus in family Costaceae.
Figure 1. Maximum likelihood tree based on the sequences of 25 chloroplast genomes. The bootstrap values were based on 1,000 replications and are indicated next to the branches. Accession numbers: Alpinia zerumbet JX088668, Alpinia oxyphylla KY985237, Amomum kravanh MF991963, Amomum compactum MG000589, Canna indica KF601570, Chamaecostus acaulis MH603404, Cheilocostus speciosus MH603405, Costus dubius MH603406, Costus gabonensis MH603407, Costus osae MH603408, Costus pictus MH603409, Costus pulverulentus KF601573, Curcuma roscoeana KF601574, Curcuma flaviflora KR967361, Dimerocostus strobilaceus MH603413, Musa acuminata subsp. malaccensis HF677508, Musa textilis KF601567, Monocostus uniflorus 1 KF601572, Monocostus uniflorus 2 MH603429, Musa balbisiana KT595228, Musa itinerans KY753133, Musella lasiocarpa KY807173, Tapeinochilos ananassae MH603446, and Zingiber spectabile JX088661.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Chaisson MJ, Tesler G. 2012. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinform. 13:238.
- Denisov G, Walenz B, Halpern AL, Miller J, Axelrod N, Levy S, Sutton G. 2008. Consensus generation and variant detection by celera assembler. Bioinformatics. 24:1035–1040.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30:772–780.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28:1647–1649.
- Kumar S, Stecher G, Tamura K. 2016. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 33:1870–1874.
- Li X, Hu Z, Lin X, Li Q, Gao H, Luo G, Chen S. 2012. High-throughput pyrosequencing of the complete chloroplast genome of Magnolia officinalis and its application in species identification. Acta Pharmaceutica Sinica. 47:124–130.
- Lohse M, Drechsel O, Kahlau S, Bock R. 2013. Organellar Genome DRAW—a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41:W575–W581.
- Lowe TM, Chan PP. 2016. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44:W54–W57.
- Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, He G, Chen Y, Pan Q, Liu Y. 2012. SOAPdenovo2: an empirically improved memory-efficient short-end de novo assembler. Gigascience. 1:18.
- Wu D, Larsen K. 2000. Costaceae vol 24. Flora of China. Beijing: Science Press; p. 320–321.
- Wu D, Liu N, Ye Y. 2016. The Zingiberaceous resources in China. Huazhong University of Science and Technology. Wuhan: University Press; p. 6–7.
- Wyman SK, Jansen RK, Boore JL. 2004. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 20:3252–3255.