
Abstract
The cultivated apple (Malus domestica Borkh.) is one of the most important crop fruits with high-economic values in the world. In the present study, we characterized the complete chloroplast (cp) genome sequence of apple cultivar ‘Yantai Fuji 8’. The complete cp genome is 160,062 bp in length with a typical quadripartite structure. A total of 112 unique genes were found in the newly sequenced genome, including 78 protein-coding, 30 tRNA, and 4 rRNA genes. Of these, 7 protein-coding genes, 7 tRNA genes, and all 4 rRNA genes are duplicated in the inverted regions. A maximum likelihood phylogenetic tree was reconstructed using the full length of cp genome to show the relationships among species in Rosaceae. The complete cp genome will be potential genetic resources for apple breeding programs.
The cultivated apple (Malus domestica Borkh.) is one of the most economically important fruit crops widely distributed in temperate regions. More than 7500 known apple cultivars have been documented (Janick Citation2010). However, there is still some uncertainty about the parentage of commercially imported varieties because the record of breeding is sometimes incomplete (Cornille et al. Citation2014). In recent decades, chloroplast DNA has been used to address questions concerning maternal lineage (Wu et al. Citation2010; Daniell et al. Citation2016) in some species. In addition, phylogenomic analysis using complete cp genome sequences provides new insight into the evolution and domestication of some important fruit trees (Nikiforova et al. Citation2013; Jose et al. Citation2015). In this study, we determined the complete chloroplast genome of the economically important apple variety ‘Yantai Fuji8’ in Shandong, China. The annotated cp genome sequence was submitted to GenBank under accession number MH595623.
Total genomic DNA was extracted from mature leaves sampled from an experimental orchard in Nanjing Forestry University (116.355, 40.013) using modified CTAB protocol. A voucher specimen was deposited in Nanjing Forestry University. Purified DNA was fragmented and used to construct paired-end libraries with average inserted-size 500 bp. At least, DNA libraries were sequenced on Illumina HiSeq X platform (Illumina, San Diego, CA) for paired-end 150 bp reads. After filtered and trimmed by fastp program (Chen et al. Citation2018), clean reads were obtained and subsequently, the high-quality paired-end reads were used to de novo assemble the complete cp genome with chloroExtractor (Ankenbrand et al. Citation2018). Genome annotation was performed using the online program GeSeq (Tillich et al. Citation2017) with reference to the Malus baccata and Malus doumeri cp genomes previously reported (Zhang et al. Citation2017). The annotation result was inspected using Geneious 8.0.4 (Kearse et al. Citation2012) and adjusted manually as needed. The cp genome map () was drawn using the online tool OGDRAW (Lohse et al. Citation2007).
Figure 1. Chloroplast genome maps of ‘Yantai Fuji 8’. Genes drawn outside the outer circle are transcribed clockwise, and those inside are transcribed counter-clockwise. Genes belonging to different functional groups are color-coded.
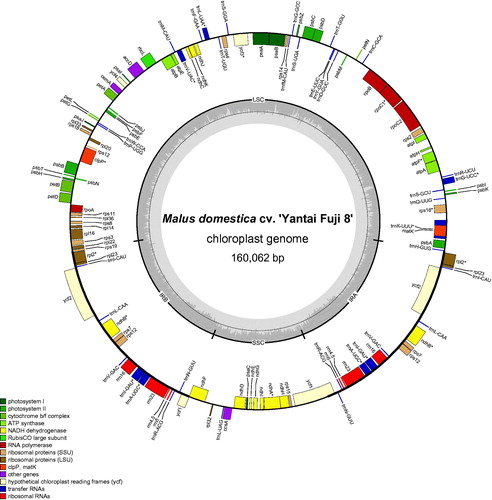
The complete cp genome of ‘Yantai Fuji’ was 160,062 bp in size, which is slightly shorter than the already reported cp genome of ‘Golden delicious’. It exhibited a typical quadripartite structure, consisting of a pair of inverted repeats (IRs, 26,352 bp) separated by a large single copy (LSC, 88,184 bp) and a small single copy (SSC, 19,174 bp). There are 112 unique genes to be found, including 78 protein-coding genes, 30 tRNAs and 4 rRNAs. Moreover, 6 protein-coding genes (rpl2, ycf2, ndhB, rps7, rps12, rpl23), 7 tRNA genes (trnI-CAU, trnL-CAA, trnV-GAC, trnI-GAU, trnA-UGC, trnN-GUU, trnR-ACG), and all rRNA genes (5S, 4.5S, 23S, 16S) are located at the IR regions. Ten of the protein-coding genes and six of the tRNA genes contain introns, 14 of which contain a single intron, whereas two (ycf3, clpP) have two introns. In particular, the rps12 is a trans-spliced gene, whose first exon is located at LSC while the second and third exons reside in IRs.
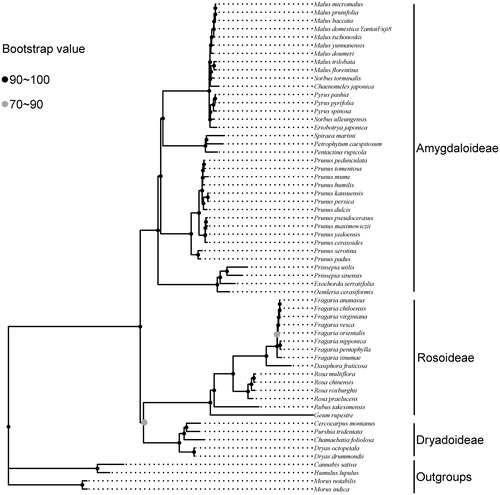
Forty-six species from Rosaceae and 4 species from Moraceae and Cannabaceae as outgroups were used for the phylogenetic reconstruction. The complete cp genomes were aligned using MAFFT (Katoh and Toh Citation2010). A maximum likelihood phylogenetic tree was inferred using IQ-TREE (Nguyen et al. Citation2015), with branch support estimated using 1000 replicates. The optimal substitution models were identified using the ModelFinder (Kalyaanamoorthy et al. Citation2017). Our result of the phylogenetic analysis () was compatible with the taxonomic treatment of Rosaceae with three subfamilies Amygdaloieae, Dryadoideae, and Rosoideae (Zhang et al. Citation2017).
Figure 2. Phylogenetic tree based on 56 complete chloroplast genome sequences of Rosaceae with maximum likelihood (ML).
Disclosure statement
The authors declare no conflict of interest.
Additional information
Funding
References
- Ankenbrand MJ, Pfaff S, Terhoeven N, Qureischi M, Gündel M, Weiß CL, Hackl T, Förster F. 2018. ChloroExtractor: extraction and assembly of the chloroplast genome from whole genome shotgun data. JOSS. 3:464.
- Chen S, Zhou Y, Chen Y, Gu J. 2018. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 34:i884–i890.
- Cornille A, Giraud T, Smulders MJM, Roldán-Ruiz I, Gladieux P. 2014. The domestication and evolutionary ecology of apples. Trends Genet. 30:57–65.
- Daniell H, Lin CS, Ming Y, Chang WJ. 2016. Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17:134.
- Janick J. 2010. The origins of fruits, fruit growing, and fruit breeding. Hoboken, New Jersey: John Wiley & Sons, Inc.
- Jose CC, Roberto A, Victoria IE, Javier T, Manuel T, Joaquin D. 2015. A phylogenetic analysis of 34 chloroplast genomes elucidates the relationships between wild and domestic species within the genus Citrus. Mol Biol Evol. 32:2015.
- Kalyaanamoorthy S, Minh BQ, Tkf W, Von HA, Jermiin LS. 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nature Methods. 14:587–589.
- Katoh K, Toh H. 2010. Parallelization of the MAFFT multiple sequence alignment program. Bioinformatics. 26:1899.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28:1647–1649.
- Lohse M, Drechsel O, Bock R. 2007. OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr Genet. 52:267–274.
- Nguyen LT, Schmidt HA, Von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32:268–274.
- Nikiforova SV, Duccio C, Riccardo V, Vadim G. 2013. Phylogenetic analysis of 47 chloroplast genomes clarifies the contribution of wild species to the domesticated apple maternal line. Mol Biol Evol. 30:1751–1760.
- Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, Greiner S. 2017. GeSeq – versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45:W6–W11. (Web Server issue).
- Wu FH, Chan MT, Liao DC, Hsu CT, Lee YW, Daniell H, Duvall MR, Lin CS. 2010. Complete chloroplast genome of Oncidium Gower Ramsey and evaluation of molecular markers for identification and breeding in Oncidiinae. BMC Plant Biol. 10:68.
- Zhang SD, Jin JJ, Chen SY, Chase MW, Soltis DE, Li HT, Yang JB, Li DZ, Yi TS. 2017. Diversification of Rosaceae since the late cretaceous based on plastid phylogenomics. New Phytol. 214:1355.