
Abstract
Carpinus oblongifolia (Betulaceae) is a critically endangered species endemic to Jiangsu. In this study, the complete chloroplast (cp) genome of C. oblongifolia was assembled based on next-generation sequencing. The cp genome was 159,086 bp in length, including a large single-copy (LSC) region of 88,190 bp, a small single-copy (SSC) region of 18,765 bp and a pair of inverted repeats (IRs) of 26,066 and 26,067 bp, respectively. The genome contained 124 genes, including 85 protein-coding genes, 30 tRNA genes, and 8 ribosomal RNA genes. The overall GC content of the chloroplast genome was 36.49%. A phylogenetic analysis demonstrated a close relationship between C. oblongifolia and Ostrya rehderiana.
Carpinus oblongifolia is one of the tall deciduous trees (10–15 m) within the family Betulaceae, and only distributed in Mountain Baohua, Jiangsu, China. This species is listed as critically endangered species in the China Species Red List (Wang and Xie Citation2004). Therefore, it is urgent to take effective measures to protect this endangered species. In the last decade, next-generation high-throughput sequencing technologies have allowed producing abundant genomic resources of our interested species in a rapid and cost-effective way (Hao et al. Citation2011). Here, we determined the complete chloroplast genome sequence of C. oblongifolia (GenBank: MG720817), which might provide us molecular information with a better insight for conservation and restoration efforts.
A fresh leaf was collected from a mature individual of C. oblongifolia in Mountain Baohua, Jiangsu, China (32.12°N, 119.06°E), then it was dried using silica gel. Genomic DNA was isolated using the modified CTAB method (Doyle and Doyle Citation1987) and kept in the Institute of Molecular Ecology of Lanzhou University. The whole-genome sequencing was conducted with 150 bp pair-end reads on the Illumina Hiseq Platform (Illumina, San Diego, CA). In total, 58.2 million high-quality base pairs of sequence data were obtained and used for the cp genome assembly using the Fast-Plast v1.2.8 pipeline (https://github.com/mrmckain/Fast-Plast) (McKain and Wilson Citation2018). Annotation was performed with a newly developed command-line application called Plann (Plastome Annotator), which is suitable for plastomes annotation with a well-annotated sequenced relative (Huang and Cronk Citation2015). Then, we corrected the annotation and generated the plastid genome map with Geneious (Kearse et al. Citation2012). We also constructed the phylogenetic trees with neighbor-joining (NJ, Phylip version 3.696, Felsenstein Citation2005), the maximum likelihood (ML, RaxML version 8, Stamatakis Citation2014) and bayesian analysis (BI, MrBayes version 3, Ronquist and Huelsenbeck Citation2003) methods based on the alignments created by the MAFFT (Katoh and Standley Citation2013) within the 9 species plastid genomes.
The complete chloroplast genome of C. oblongifolia is 159,086 bp in length, comprising a large single-copy (LSC) of 88,190 bp and a small single-copy (SSC) of 18,765 bp, separated by two inverted repeat (IR) regions of 52,133 bp. It contained 124 genes, included 85 protein-coding, 8 ribosomal RNA, and 30 tRNA genes. The plastome contained 95 unique genes, 14 genes duplicated in the IR regions. Among annotated genes, 10 genes contained a single intron, and 4 genes had two introns (i.e., clpP, ycf3, rps12, and rpl2). The base compositions of the whole chloroplast genome were uneven (31.32% A, 18.61% C, 17.88% G, 32.19% T), with an overall GC content of 36.49%, and the corresponding values of the LSC, SSC, and IR regions reaching 34.29%, 30.18%, and 42.48%, respectively.
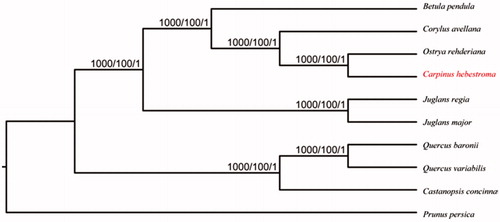
To ascertain the phylogenetic position of C. oblongifolia, we conducted a phylogenetic analysis using 10 complete chloroplast genomes (). The result indicated that C. oblongifolia was most closely related to Ostrya rehderiana. This complete chloroplast genome can be readily used for population genomic studies of C. oblongifolia, and such information would be fundamental to formulate potential new conservation and management strategies for this endangered species.
Figure 1. Phylogenetic relationships of nine Betulaceae species based on chloroplast genome sequences with Prunus persica (Rosaceae, HQ336405) as an outgroup. NJ/ML/Bayesian posterior probabilities/bootstrap values are shown at nodes. Accession numbers: Betula pendula LT855378, Corylus avellana KX822768, Ostrya rehderiana KT454094, Carpinus oblongifolia MG720817, Juglans regia NC_028617, Juglans major NC_035966, Quercus baronii KT963087, Quercus variabilis KU240009, Castanopsis concinna NC_033409.
Acknowledgments
The authors would like to thank Ying Li in Lanzhou University for the assistance in sample collection and identification.
Disclosure statement
No potential conflict of interest is reported by the authors.
Additional information
Funding
References
- Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19:11–15.
- Felsenstein J. 2005. PHYLIP (Phylogeny Inference Package) version 3.6. distributed by the author. Seattle: Department of Genome Sciences, University of Washington.
- Hao Y, Tardivo L, Tam S, Weiner E, Gebreab F, Fan C, Sahalie J. 2011. Next-generation sequencing to generate interactome datasets. Nat Methods. 8:478–480.
- Huang DI, Cronk Q. 2015. Plann: a command-line application for annotating plastome sequences. Appl Plant Sci. 3:1500026.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30:772–780.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28:1647–1649.
- McKain MR, Wilson M. 2018. Fast-plast: rapid de novo assembly and finishing for whole chloroplast genomes. Version 1.2.8.
- Ronquist F, Huelsenbeck JP. 2003. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 19:1572–1574.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30:1312–1313.
- Wang S, Xie Y. 2004. China Species Red List Vol. 1: Red List. Beijing, China: Higher Education Press.