
Abstract
Pyrus phaeocarpa Rehd. belongs to the Pyrus genus (Rosaceae) with high commercial value in China. Many trees in the wild have been destroyed affected by human activities and needed to be protection urgently. In this study, we reported the complete chloroplast genome sequence of P. phaeocarpa using Illumina sequencing technology. The complete chloroplast genome of P. phaeocarpa was 160,203 bp in length, containing a large single copy (LSC, 88,200 bp), a small single copy (SSC, 19,231 bp), and a pair of inverted repeats (IRs, 26,385 bp). The chloroplast genome GC content was 36.54%, contained 113 unique genes, including 30 tRNA, four rRNA, and 79 protein-coding genes. Phylogenetic analysis revealed that P. phaeocarpa was close to P. pashia, P. pyrifolia, and P. ussuriensis, provided strong support for the relationship of Pyrus.
Pyrus is the second most important rosaceous fruit species, origin from tertiary or possibly even more ancient periods based on paleontological data (Rubtsov Citation1944; Hummer and Janick Citation2009). Pyrus phaeocarpa is of the genus Pyrus of Rosaceae, Rosales, which is distributed among northern China (Bell et al. Citation1996). The rootstock often used as stock to graft pear cultivars as important germplasm resources for genetic breeding of pears (Gu and Stephen Citation1994). This species is hermaphrodite and has important research value in system evolution of the genus Pyrus. In recent years, some wild resources have been destroyed due to frequent human activities, understanding its genomics will contribute to the formulation of protection strategies and interspecific relationships of Pyrus.
The young leaves sample of wild P. phaeocarpa was collected from Xiangyang, Shanxi, China (112°1'6.3″E, 35°27'43.4″N), the voucher specimen (PPR001) was deposited at the herbarium of Museum of Beijing Forestry University. Total genomic DNA was extracted using CTAB method (Doyle and Doyle Citation1987) and sequenced on the Illumina Hiseq X–Ten Sequencing platform at Yuanyi, Beijing, China (http://www.ori–gene.cn/). Raw reads were quality control used the FastQC software (Andrews Citation2010), then clean reads were assembled into a single draft sequence with the reference chloroplast genome of P. hopeiensis (MF521826) (Li et al. Citation2018) using SOAPdenovo and BLAT (Kent Citation2002; Luo et al. Citation2012). The draft sequence was filled the gaps with high–quality short sequences using SOAPdenovo GapCloser software (Luo et al. Citation2012). The sequence was annotated using CpGAVAS (Liu et al. Citation2012), then manually auxiliary annotation by using DOGMA (Wyman et al. Citation2004). The P. phaeocarpa chloroplast genome sequence has been submitted to GenBank using Bankit (https://www.ncbi.nlm.nih.gov/books/NBK63590/) (accession number: MK488091).
The complete chloroplast genome of P. phaeocarpa was 160,203 bp in length with 36.54% overall GC content, consist of a small single copy (SSC) region of 19,231 bp, a large single copy (LSC) region of 88,200 bp, and a pair repeat regions of inverted repeat (IRa and IRb) of 26,385 bp by each. The chloroplast genome encodes 113 unique genes, including 79 protein–coding genes, 30 tRNA genes, and four rRNA genes.
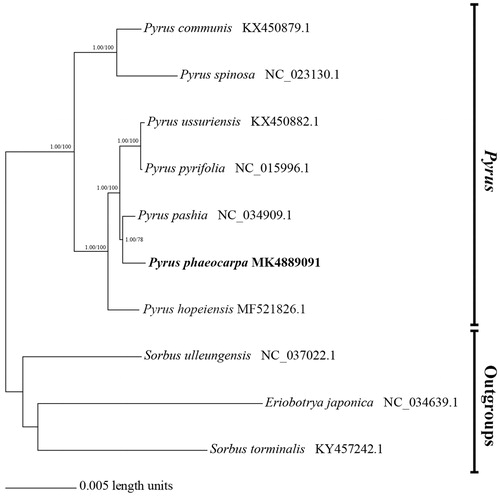
The phylogenetic relationship of P. phaeocarpa was determined based on six representative species of Pyrus and three outgroups (Sorbus ulleungensis, Eriobotrya japonica, and Sorbus torminalis) using PAUP* 4.10 (Swofford Citation2003) by Maximum Parsimony (MP). Bayesian inference (BI) was performed with Mrbayes v3.2.3 (Ronquist et al. Citation2012). The phylogenetic tree showed that P. phaeocarpa was close to P. pashia, P. pyrifolia, and P. ussuriensis (), which is coherent with a previous report that considered P. phaeocarpa might have developed from the hybridization of P. betulaefolia, P. pyrifolia and P. ussuriensis (Jiang et al. Citation2016) may have a closer evolutionary relationship.
Figure 1. Phylogenetic relationships of the 10 species inferred from Maximum Parsimony (MP) and Bayesian inference (BI) based on complete genome sequences. MP bootstrap values/pp values for Bayesian analysis are shown at each node.
Disclosure statement
The authors have declared that no competing interests exist.
Additional information
Funding
Related Research Data
References
- Andrews S. 2010. FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- Bell RL, Quamme HA, Layne REC, Skirvin RM. 1996. Pears. In: Janick J, Moore JN, editors. Fruit breeding, tree and tropical fruits. New Jersey: John Wiley & Sons; p. 441–451.
- Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19:11–15.
- Gu CZ, Stephen AS. 1994. PYRUS Linnaeus, Sp. Pl. 1: 479. 1753. In: Wu ZY, Raven P, editors. Flora of China. Beijing: Science Press; St. Louis: Missouri Botanical Garden Press; p. 173–179.
- Hummer KE, Janick J. 2009. Rosaceae: taxonomy, economic importance, genomics. In: Folta KM, Gardiner SE, editors. Genetics and genomics of Rosaceae. New York: Springer; p. 1–17.
- Jiang S, Zheng XY, Yu PY, Yue XY, Ahmed M, Cai DY, Teng YW. 2016. Primitive genepools of Asian pears and their complex hybrid origins inferred from fluorescent sequence–specific amplification polymorphism (SSAP) markers based on LTR retrotransposons. PloS One. 11:e0149192.
- Kent WJ. 2002. BLAT-the BLAST-like alignment tool . Genome Res. 12:656–664.
- Li W, Lu Y, Xie X, Li B, Han Y, Sun T, Xian Y, Yang H, Liu K. 2018. Development of chloroplast genomic resources for Pyrus hopeiensis (Rosaceae). Conservation Genet Resour. 10:511–513.
- Liu C, Shi LC, Zhu YJ, Chen HM, Zhang JH, Lin XH, Guan XJ. 2012. CpGAVAS, an integrated web server for the annotation, visualization, analysis, and GenBank submission of completely sequenced chloroplast genome sequences. BMC Genomics. 13:715.
- Luo RB, Liu BH, Xie YL, Li ZY, Huang WH, Yuan JY, He GZ, Chen YX, Pan Q, Liu YJ, et al. 2012. SOAPdenovo2: an empirically improved memory–efcient short–read de novo assembler. Gigascience. 1:18.
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61:539–542.
- Rubtsov GA. 1944. Geographical distribution of the genus Pyrus and trends and factors in its evolution. Am Nat. 78:358–366.
- Swofford DL. 2003. PAUP*. Phylogenetic Analysis Using Parsimony (and Other Methods). Version 4.
- Wyman SK, Jansen RK, Boore JL. 2004. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 20:3252–3255.