
Abstract
Fortunearia sinensis Rehder & E.H.Wilson, the only species of monotypic genus Fortunearia (Hamamelidaceae), is a native rare and vulnerable deciduous tree in China. In this study, the complete plastid genome of F. sinensis was determined using the Illumina paired-end sequencing data. The complete plastid genome of F. sinensis is 1,59,441 bp in length, consisting of a pair of inverted repeats (IR) regions (26,268 bp), a large-single copy (LSC) region (88,124 bp), and a small-single copy (SSC) region (18,781 bp). The plastid genome encodes 112 unique genes, including 80 protein-coding genes, 28 tRNA genes, and 4 rRNA genes. The phylogenetic analysis demonstrates a close relationship between F. sinensis and Sinowilsonia henryi.
Fortunearia sinensis Rehder & E.H. Wilson is the only species of the monotypic genus Fortunearia in Hamamelidaceae. This species is an endemic medicinal plant in China, since its leaves contain bergenin, fortunearioside, and other medicinal ingredients (Wang Citation1996). In addition, F. sinensis is economically important because of its high oil content in seeds (Jia and Zhou Citation1987). It has been assessed as a vulnerable species in China (Qin et al. Citation2017). In this study, we characterized the complete plastid genome sequence of F. sinensis based on Illumina paired-end sequencing for further genetic studies, conservation, and utilization of the endangered and rare species.
Fresh leaves of F. sinensis were collected from Wuhan Botanical Garden and dried with silica gel. The voucher specimen (voucher number: wh271) was deposited in the Herbarium of South China Botanical Garden (IBSC). Total genomic DNA was extracted using the modified CTAB method (Doyle and Doyle Citation1987). The prepared Illumina paired-end library with an average insert size of 270 bp was sequenced on a Hiseq X Ten platform. After quality filtering and trimming, c. 3.0 Gb clean data was assembled with a reference plastid genome of Sinowilsonia henryi Hemsl. (GenBank accession number: NC_036069) using NOVOPlasty (Dierckxsens et al. Citation2017). We remapped the clean reads to the resulting circular sequence in BWA version 0.7.17 (Li Citation2013), and manually identified and fixed single nucleotide and small structural errors using Geneious version R11.1.5 (Biomatters Ltd., Auckland, New Zealand). The annotation of the plastome was performed through the online programme GeSeq – Annotation of organellar genomes (Tillich et al. Citation2017) with necessary manual adjustment in Geneious.
The plastome of F. sinensis (GenBank accession number MK533616) is 159,441 bp in length. The genome shows a typical quadripartite structure, containing two copies of inverted repeat (IR) regions (26,268 bp), a large-single copy (LSC) region (88,124 bp), and a small-single copy (SSC) region (18,781 bp). Its overall GC content is 38.1%, while the corresponding values of the LSC, SSC, and IR regions are 36.3, 32.9, and 43.1%, respectively. A total of 112 unique genes were annotated, containing 80 protein-coding genes, 28 tRNA genes, and 4 rRNA genes ().
Table 1. List of 112 unique genes in the plastome of Fortunearia sinensis.
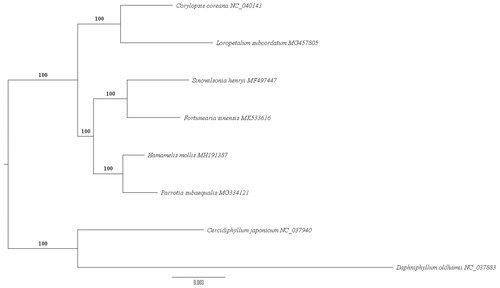
Seven published plastid genome sequences, belonging to five species in Hamamelidaceae and two outgroups (Daphniphyllum oldhamii and Cercidiphyllum japonicum), were downloaded from GenBank for inferring the phylogenetic position of F. sinensis. A total of 79 common protein-coding genes of all eight plastomes were aligned separately using MAFFT (Katoh and Standley Citation2013) and concatenated into a single matrix in Geneious. The maximum likelihood (ML) tree based on the combined matrix was reconstructed using RAxML version 8.2.9 (Stamatakis Citation2014) with a basis of 1000 bootstrap replicates (). The result showed the species of Hamamelidaceae formed a monophyletic clade with 100% bootstrap value. As expected, F. sinensis and S. henryi were recovered as sister with high support (100%), which agreed with the phylogenetic result of Li and Bogle (Citation2001).
Figure 1. Phylogenetic relationships of Hamamelidaceae inferred from maximum likelihood method based on 79 common protein-coding genes. Daphniphyllum oldhamii and Cercidiphyllum japonicum were used as outgroups. The node labels indicate the ML bootstrap (1000 replicates) support values.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45:e18.
- Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19:11–15.
- Jia LZ, Zhou J. 1987. Fat-bearing plants in China. Beijing, China: Science Press.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30:772–780.
- Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. 1303:3997.
- Li JH, Bogle AL. 2001. A new suprageneric classification system of the Hamamelidoideae based on morphology and sequences of nuclear and chloroplast DNA. Harv Papers Bot. 5:499–515.
- Qin HN, Yang Y, Dong SY, He Q, Jia Y, Zhao LN, Yu SX, LHY, Liu B, Yan YH, et al. 2017. Threatened species list of China’s higher plants. Biodivers Sci. 25:696–744.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30:1312–1313.
- Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, Greiner S. 2017. GeSeq - versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45:W6–W11.
- Wang JS. 1996. Hamamelidaceae denoted. Plants. 5:32–34.