
Abstract
Hedysarum taipeicum is a threatened perennial herb with medicinal value, which is an endemic species only distributed in Taibai Mountain, Shaanxi Province, China. The complete chloroplast genome of H. taipeicum was sequenced using the Illumina Hiseq 4000 platform. Its genome is relatively short (126,699 bp) because it lacks an Inverted Repeat (IR) region. The genome contains 110 genes, including 76 protein-coding genes, 30 transfer RNA genes (tRNA), 4 ribosomal RNA genes (rRNA). The overall GC content of the whole genome is 35.1%. Phylogenetic analysis based on protein sequences of 17 chloroplast genomes indicates that H. taipeicum is closely related to Glycyrrhiza glabra in Leguminosae family.
Hedysarum taipeicum is a perennial herb belonging to genus Hedysarum of the family Leguminosae (Qin et al. Citation2017). The genus Hedysarum (sweetvetch) consists of about 200 species of annual or perennial herbs spread over Asia, Eroupe, North America, and North Africa (Duan et al. Citation2015). Taibai sweetvetch (H. taipeicum) is endemic to Taibai Mountains in Shaanxi Province and is born on gravel slopes and watershed grasslands from 1500–3300 m (Tian and Liu Citation1996; Dong et al. Citation2013). Despite this species’ traditional Chinese medicine value (Dong et al. Citation2013) and being endangered in Qinling mountains (Qin et al. Citation2017), existing genomic resources for H. taipeicum, and genetic studies have been limited for Taibai sweetvetch. Here we hvae reported first complete chloroplast genomes of H. taipeicum based on Illumina Hiseq 4000 pair-end sequencing data.
The plant material of H. taipeicum was sampled from Taibai Mountain, Shaanxi Province, China. The voucher specimen is kept at the Evolutionary Botany Laboratory, College of Life Sciences, Northwest University (Xi’an, Shaanxi, China). Total genomic DNA was extracted from leaf tissue using the Plant Genomic DNA kit (Tiangen Biotech, Beijing, China). The whole-genome sequencing was conducted with 350 bp pair-end reads on the IlluminaHiseq 4000 platform (Illumina, San Diego, CA) by Novogene, Beijing, China.
After trimming, the high-quality paired-end reads were assembled with the program MITObim v1.7 (Hahn et al. Citation2013) using the Astragalus mongholicus chloroplast genome sequence as a reference (GenBank accession number KU666554) (Lei et al. Citation2016). Annotations were performed using the online program Dual Organellar Genome Annotator (DOGMA) (Wyman et al. Citation2004). We corrected annotations using Geneious (Kearse et al. Citation2012). A map of the genome was generated using OGDRAW (Lohse et al. Citation2013). The chloroplast genome sequence was submitted to GenBank (accession number MK426698).
The cp genome of H. taipeicum is 126,699 bp in length and lacks an IR region. Its 110 genes include 76 protein-coding genes, 30 transfer RNA genes, and 4 ribosomal RNA genes. Among these genes, 16 genes (atpF, ndhA, ndhB, petB, petD, rpl2, rpl16, rpoC1, rps12, clpP, trnE-UUC, trnA-UGC, trnG-UCC, trnC-ACA, trnK-UUU, and trnL-UAA) have one intron, and one gene (ycf3) has two introns. Overall GC content is 35.1%.
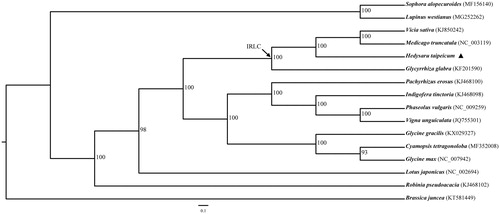
To conduct phylogenetic analysis, we downloaded complete chloroplast genome sequences of 15 Leguminosae species from NCBI, and Brassica juncea was included in the analysis as outgroup. The protein sequences of these 16 sequences were extracted using Geneious (Kearse et al. Citation2012) and aligned using MAFFT v7.0.0 (Katoh and Standley Citation2013). The phylogenetic relationships analysis was inferred using the Maximum likelihood (ML) method based on protein-coding genes, which was performed using RAxML (Stamatakis Citation2006). The local bootstrap probability of each branch was calculated by 1000 replications. The phylogenetic tree showed that H. taipeicum was most closely related to Glycyrrhiza glabra with 100% bootstrap support ().
Figure 1. Maximum likelihood (ML) phylogenetic tree based on protein sequences of 17 chloroplast genomes. The accession numbers showed in the figure, and the triangle indicates Hedysarum Taipeicum. IRLC indicateds the inverted-repeat-lacking clade.
Disclosure statement
The authors report no conflicts of interest.
Additional information
Funding
References
- Dong Y, Tang D, Zhang N, Li Y, Zhang C, Li L, Li M. 2013. Phytochemicals and biological studies of plants in genus Hedysarum. Chem Cent J. 7:124.
- Duan L, Wen J, Yang X, Liu P-L, Arslan E, Ertuğrul K, Chang Z-Y. 2015. Phylogeny of Hedysarum and tribe Hedysareae (Leguminosae: Papilionoideae) inferred from sequence data of ITS, matK, trnL-F and psbA-trnH. Taxon. 64:49–64.
- Hahn C, Bachmann L, Chevreux B. 2013. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads–a baiting and iterative mapping approach. Nucleic Acids Res. 41:e129.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30:772–780.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28:1647–1649.
- Lei W, Ni D, Wang Y, Shao J, Wang X, Yang D, Wang J, Chen H, Liu C. 2016. Intraspecific and heteroplasmic variations, gene losses and inversions in the chloroplast genome of Astragalus membranaceus. Sci Rep. 6:21669.
- Lohse M, Drechsel O, Kahlau S, Bock R. 2013. OrganellarGenomeDRAW-a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41:W575–W581.
- Qin H, Yang Y, Dong S, He Q, Jia Y, Zhao L, Yu S, Liu H, Liu B, Yan Y, et al. 2017. Threatened species list of China’s higher plants. Biodivers Sci. 25:696–744.
- Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 22:2688–2690.
- Tian HY, Liu QG. 1996. A study on the chemical constituents of Hedysarum Taipeicum (Hand-Mazz) K T Fu. J Southwest Nationalities College Nat Sci. 22:182–184.
- Wyman SK, Jansen RK, Boore JL. 2004. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 20:3252–3255.