
Abstract
The complete chloroplast (cp) genome of Rosa berberifolia was assembled from high-throughput Illumina sequencing data. The cp genome of R. berberifolia was 156,886 bp in length with a typical quadripartite circular, containing a pair of inverted repeats (26,047 bp) separated by a large (86,045 bp), and small (18,747 bp) single-copy regions. It contains 129 genes, including 84 protein-coding genes, 8 ribosomal RNA genes, 37 transfer RNA genes, and 1 pseudogene (ycf1). The overall GC content of the whole cp genome was 37.2%. Phylogenetic analysis based on the complete chloroplast genomes suggested that R. berberifolia was sister to R. persica.
Rosa berberifolia Pallas (Rosaceae) is a shrub species in the family Rosaceae with important ecological and ornamental values. It can grow well in the Steppes and desert regions of China, Kazakhstan, and Russia (Ku and Robertson Citation2003). It is considered as a potential source for resistance against drought stress (Hui et al. Citation2014). It is a desert ornamental plant which has a large, yellow flower with adark-colored central zone. In addition, R. berberifolia is an anomalous species in the genus Rosa and was previously placed in a separate genus Hulthemia. Its distinctive characteristics include a simple leaf without stipules (most rose leaves are pinnate with 3–7 leaflets, and have stipules) and petals with a purple-red spot at the base. To provide genomic resources for investigating the evolution of R. berberifolia, the complete chloroplast (cp) genome of this species was assembled and characterized using high-throughput sequencing data.
The fresh leaves of R. berberifolia were collected from Manas County, Xinjiang Uygur Autonomous Region (44°06′13″N, 86°21′07″E, 864 m). The voucher specimen (16CS13205) was deposited in Herbarium, Kunming Institute of Botany, CAS (KUN). Genomic DNA was isolated using the modified CTAB method (Doyle Citation1987) and used for sequencing on the Illumina HiSeq 2500 Platform by Novogene Bioinformatics Technology Co., Ltd. (Beijing, China). The cp genome was de novo assembled using the GetOrganelle pipeline (https://github.com/Kinggerm/GetOrganelle). The annotation of the cp genome was conducted using Dual Organellar Genome Annotator (Wyman et al. Citation2004) with manual adjustments. The online tRNAscan-SE Search Service (Lowe and Chan Citation2016; http://lowelab.ucsc.edu/tRNAscan-SE/) was used to further confirm tRNA genes. The complete cp genome sequence of R. berberifolia was submitted to GenBank under the accession number MK423879.
The cp genome of R. berberifolia was 156,886 bp in length with a typical quadripartite structure consisting of a large single copy (LSC) of 86,045 bp, a small single copy (SSC) region of 18,747 bp, and a pair of 26,047 bp inverted repeats (IRs). The GC content of the complete cp genome was 37.2%. The genome contained 129 genes, including 84 protein-coding genes (PCGs), 37 transfer RNA (tRNA) genes, and 8 ribosomal RNA (rRNA) genes. Six coding sequences (CDSs), seven tRNA genes, and all the four rRNA genes were duplicated in the IR regions. In addition, three PCGs (clpP, rps12, and ycf3) had two introns, eight PCGs (ndhA, ndhB, petB, petD, rpl16, rpl2, rpoC1, and rps16) and six tRNA genes (trnA-UGC, trnG-UCC, trnI-GAU, trnK-UUU, trnL-UAA, and trnV-UAC) contained one intron. The genome structure, GC content, and gene locations of the cp genome of R. berberifolia were almost identical to those of the other species of Rosa (Jian et al. Citation2017, Citation2018).
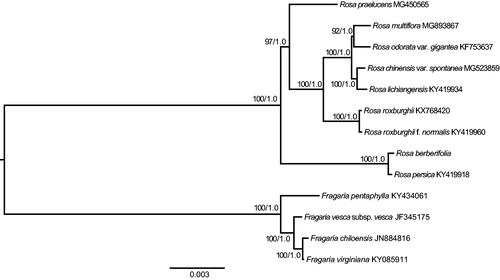
Phylogenomic analysis based on the complete cp genome was performed with the maximum likelihood (ML) and Bayesian inference (BI) methods (Ronquist and Huelsenbeck Citation2003; Stamatakis Citation2014). Four species from Fragaria (F. pentaphylla, F. vesca subsp. vesca, F. chiloensis and F. virginiana) were used as the outgroups suggested in previous phylogenetic studies (Potter et al. Citation2007; Zhang et al. Citation2017). The GenBank accession numbers of all the species used for phylogenomic analysis are provided in . The complete cp genome sequences were aligned using MAFFT version 7.0 (Katoh and Standley Citation2013). The ML and BI analyses generated the same tree topology (). Rosa berberifolia and R. persica formed a monophyletic group sister to the rest of the species of Rosa. The R. berberifolia cp genome reported in this study may provide useful resources for the development of ornamental and ecological value as well as robust phylogenetic study at a deep level of Rosa in the future.
Figure 1. The maximum likelihood (ML) tree of Rosa inferred from the complete chloroplast genome sequences. Numbers at nodes correspond to ML bootstrap percentages (1000 replicates) and Bayesian inference (BI) posterior probabilities.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Doyle JJ. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19:11–15.
- Hui JA, Zhang X, Wang SM. 2014. Microstructural characteristics of wild Hulthemia berberifolia in Xinjiang. Jiangsu Agr Sci. 42:126–127.
- Jian HY, Zhang SD, Zhang T, Qiu XQ, Yan HJ, Li SB, Wang QG, Tang KX. 2017. Characterization of the complete chloroplast genome of a Critically Endangered decaploid rose species, Rosa praelucens (Rosaceae). Conservation Genet Resour. 10:851–854.
- Jian HY, Zhang YH, Yan HJ, Qiu XQ, Wang QG, Li SB, Zhang SD. 2018. The complete chloroplast genome of a key ancestor of modern roses, Rosa chinensis var. spontanea, and a comparison with congeneric species. Molecules. 23:389–401.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30:772–780.
- Ku TC, Robertson KR. 2003. Rosa (Rosaceae). In: Wu ZY, Raven PH, Hong DY, editors. Flora of China, vol 9. St. Louis and Beijing: Missouri Botanical Garden and Science Press; p. 339–381.
- Lowe TM, Chan PP. 2016. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44:W54–W57.
- Potter D, Eriksson T, Evans RC, Oh S, Smedmark JEE, Morgan DR, Kerr M, Robertson KR, Arsenault M, Dickinson TA, et al. 2007. Phylogeny and classification of Rosaceae. Plant Syst Evol. 266:5–43.
- Ronquist F, Huelsenbeck JP. 2003. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 19:1572–1574.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30:1312–1313.
- Wyman SK, Jansen RK, Boore JL. 2004. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 20:3252–3255.
- Zhang SD, Jin JJ, Chen SY, Chase MW, Soltis DE, Li HT, Yang JB, Li DZ, Yi TS. 2017. Diversification of Rosaceae since the late cretaceous based on plastid phylogenomics. New Phytol. 214:1355–1367.