
Abstract
The complete mitogenome of Sydowia polyspora NIE441 (KCTC No. 56410) isolated from alpine conifer Abies nephrolepis is determined by Illumina Hiseq4000 platform in this study. Total length of the sequenced mitogenome is 37,422 bp with 29.28% G + C content. A total of 43 genes were predicted in this mitogenome: 15 protein coding genes, 2 rRNAs, and 26 tRNAs.
Sydowia polyspora (Bref. & Tavel) E. Müll. belongs to Dothideomycetes and was found in some conifers species. In this study, S. polyspora strain NIE441 (KCTC No. 56410) was isolated from healthy needle leaf as endophytic fungi of Abies nephrolepis (Trautv.) Maxim. distributed in Mt. Jeombong (38°02′04′′N, 128°26′19′′E) of Korea and was deposited in Korean Collection for Type Cultures (KCTC). Genomic DNA was extracted from S. polyspora NIE441 mycelium using Qiagen DNeasy DNA prep kit (Qiagen, Hilden, Germany). Libraries were constructed using Truseq Nano DNA library (Illumina Inc., San Diego, CA) and sequenced on Illumina HiSeq4000 Platform (Illumina Inc., San Diego, CA) with 101 paired-end reads.
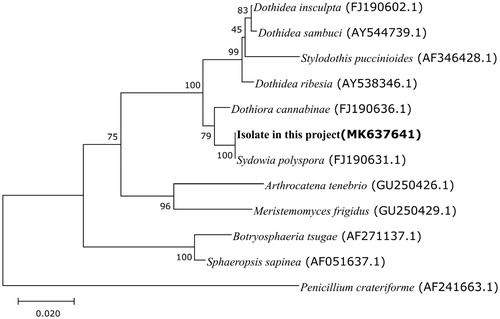
Raw reads were de novo assembled using the Ray v2.3.1. (Boisvert et al. Citation2010) and SPAdesv.3.11.1 (Bankevich et al. Citation2012) programs. In order to confirm that contigs with high coverage (1000 folded) were originated from a mitochondrial genome, BLAST-searches were performed against mitochondrial sequences from GenBank. Contigs related to mitochondrial genome sequences were extracted and merged using in-house scripts. Gene prediction and annotation was performed using MITOS (Bernt et al. Citation2013). For the construction of the phylogenetic tree with mitochondrial small subunit ribosomal RNA, 20 sequences retrieved from GenBank database were used. Nucleotide sequences were aligned using the MUSCLE algorithm in MEGA7 (Kumar et al. Citation2016). Tree-building was performed using the neighbor-joining method (Saitou and Nei Citation1987) with Kimura 2-parameter (Kimura Citation1980) as a distance model (). The genome consists of a circular chromosome of 37,422 bp with 29.28% G + C content (Genbank accession number: MK637641). A total of 43 genes were predicted in this mitochondrial genome, 15 of which are protein-coding genes. Most of the protein-coding genes were the conserved mitochondrial proteins for oxidative phosphorylation containing cox1, cox2, cox3, nad1, nad2, nad3, nad4, nad4L, nad5, nad6, atp6, atp8, and atp9. The genome also has 2 rRNAs (rrnS and rrnL) and 26 tRNAs.
Figure 1. Phylogenetic tree of small subunit ribosomal RNAs with neighbor-joining approach. Numbers above branches show maximum-likelihood bootstrap supports from 1000 nonparametric replicates. The tree was rooted by Penicillium crateriforme (AF241663.1). The scale represents the number of substitutions per site. All positions containing gaps and missing data were eliminated. There were a total of 452 positions in the final dataset.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Additional information
Funding
References
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19:455–477.
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogen Evol. 69:313–319.
- Boisvert S, Laviolette F, Corbeil J. 2010. Ray: simultaneous assembly of reads from a mix of high-throughput sequencing technologies. J Comput Biol. 17:1519–1533.
- Kimura M. 1980. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 16:111–120.
- Kumar S, Stecher G, Tamura K. 2016. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol. 33:1870–1874.
- Saitou N, Nei M. 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 4:406–425.