
Abstract
In this study, the complete chloroplast genome of Pholidota yunnanensis is presented, which represents first complete plastid genome of the genus Pholidota in the subtribe Coelogyninae. The chloroplast genome size is 159,729 bp, including a GC content of 37.3% and 135 genes (89 protein-coding genes, 38 tRNA genes, 8 rRNA genes). The genome structure is typical quadripartite, consisting of a pair of inverted repeat regions (26,638 bp) separated by a large single-copy region (LSC, 87,610 bp) and a small single-copy region (SSC, 18,843 bp). Phylogenetic analysis among 15 species based on cp genomes recovered a well-supported phylogenetic tree and indicated a close relationship between Pholidota yunnanensis and Pleione bulbocodioides.
Phodidota yunnanensis Rolfe (Orchidaceae: Coelogyninae), an Endangered and well-known traditional Chinese medicine species for use in clearing heat, purging lung and stasis, is mainly distributed in southwest China and adjacent northern Vietnam. As one of the national key protected species in China, it has been categorized as ‘Near Threatened’ (NT) by the International Union for Conservation of Nature (IUCN) and ranked in the second-class category of the Convention on International Trade in Endangered Species (CITES).
Herein, we report the complete chloroplast genome of P. yunnanensis, aiming at evaluating the utilities of chloroplast genome in the reconstruction of the phylogenetic position of the species in subtribe Coelogyninae (Orchidaceae), thus providing useful information for evolutionary dynamics and conservation studies.
Leaf samples of P. yunnanensis were obtained from Wenshan, Yunnan, China (N 23°12′, E 104°70′) and specimens were deposited in the National Orchid Conservation Center herbarium (NOCC; specimen code: Z.J.Liu 7674). Total genomic DNA was extracted using a modified CTAB method (Doyle and Doyle Citation1987). Next-generation sequencing was performed in Beijing Genomics Institute using the Illumina Hiseq 2000. In total, 15.47 Gb of clean data were generated for assembling the complete plastome using the GetOrganelle 5.1.0 (Jin et al. Citation2018).
The plastid genome sequence of Pleione bulbocodioides (KY849819.1) and Pleione formosana (MK361027.1) were used as references to annotate the plastome using software PGA (Qu et al. Citation2019) program. Geneious R11 (Kearse et al. Citation2012) was used to check the accuracy of the assembly and to adjust the start/stop codons and intron/exon boundaries of the annotation. We finally submitted the annotated plastome to GenBank (Accession number MN879405).
The complete chloroplast genome is 159,729 bp in length consisting of two inverted repeats (IR = 26,638 bp), one small single-copy (SSC = 18,843 bp), and one large single-copy (LSC = 87,610 bp) with a typical quadripartite structure. The A, T, C, G base composition of the genome is 30.9, 31.8, 18.9, and 18.4%, respectively. There were a total of 135 unique genes, including 89 protein-coding genes, 38 tRNA genes, and 8 rRNA genes.
To reveal the phylogenetic position of P. yunnanensis within the subfamily Epidendroideae, we performed the phylogenetic analysis using 14 complete plastid genome sequences downloaded from the GenBank, of which two species in the subfamily Orchidoideae were used as outgroups. The maximum-likelihood (ML) phylogenetic tree was built in IQ-TREE 1.6.11 with default parameters () (Nguyen et al. Citation2015). The Bayesian inference (BI) phylogenetic tree was built using MrBayes 3.2.6 () under the GTR + I+G + F model as suggested by ModelFinder (Kalyaanamoorthy et al. Citation2017; Ronquist et al. Citation2012).
Figure 1. The maximum-likelihood (ML) phylogenetic tree based on the complete chloroplast genome sequences. Numbers at the right of nodes are BPML/PP.
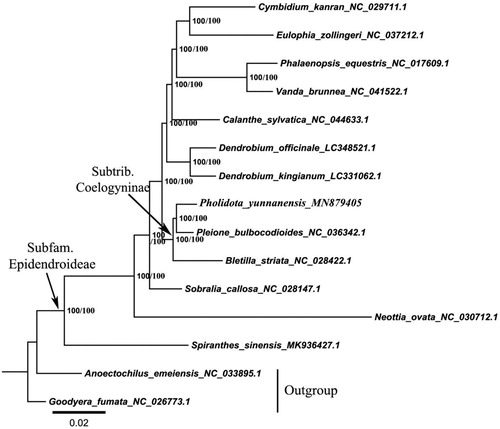
The two methods yielded highly supported and identical phylogenetic trees for all major lineages within subfamily Epidendroideae. In conclusion, the complete cp genome sequence of P. yunnanensis reported in this study will provide valuable information for identifying Pholidota species and better understand the relationships between species and plastome evolution within the tribe Coelogyninae.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19:11–15.
- Jin JJ, Yu WB, Yang JB, Song Y, Yi TS, Li DZ. 2018. GetOrganelle: a simple and fast pipeline for de novo assembly of a complete circular chloroplast genome using genome skimming data. bioRxiv. 256479. http://doi.org/10.1101/256479
- Kalyaanamoorthy S, Minh BQ, Wong TK, Haeseler AV, Jermiin LS. 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 14(6):587–589.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28(12):1647–1649.
- Nguyen LT, Schmidt HA, Haeseler AV, Minh BQ. 2015. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol Biol Evol. 32(1):268–274.
- Qu XJ, Michael JM, Li DZ, Yi TS. 2019. PGA: a software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods. 15(1):50.
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61(3):539–542.