
Abstract
Philadelphus pekinensis Rupr. is a common perennial deciduous shrub distributed in temperate China. Here, we report the complete chloroplast genome of P. pekinensis. The cp genome is totally 157,308 bp in length, including large single-copy (LSC) region of 86,457 bp, small single-copy (SSC) region of 18,735 bp, and two separated inverted regions (IRs) of 26,058 bp, respectively. It contains 134 genes, including 85 protein-coding genes, 8 rRNA genes, and 37 tRNA genes. The overall GC content is 43.1%. The phylogenetic analysis reveals the monophyly of P. pekinensis and P. calvescens, which is more related with Carpenteria californica than other species in the Hydrangeaceae family.
Philadelphus pekinensis Rupr. is famous for its beautiful, fragrantand long term flowers, making it as an ornamental shrub commonly cultivated in the botanical gardens. It grows in temperate China, Inner Mongolia, Korea, as well as Europe and North America. Here, we report the first chloroplast (cp) genome of P. pekinensis for further research on the phylogenetic and biogeographic study among its related species in the future.
Samples of P. pekinensis were collected from Yunmeng Mountain National Forest Park in Miyun District, Beijing (Voucher No.: LP161571). Total genomic DNA was extracted from silica gel dried leaves using a modified CTAB method (Doyle and Doyle Citation1987). The paired-end (2 × 150 bp) library was constructed by Illumina PE150 in Novogene Co. Ltd (Beijing, China). Totally, 2.64 Gb clean data were obtained after removing low-quality reads and adaptor sequences. The complete cp genome of P. pekinensis was assembled via GetOrganelle pipeline (available online: https://github.com/Kinggerm/GetOrganelle, JianJun et al. Citation2018), which implements four steps of recruiting plastid-like reads: (i) conducting de novo assembly using SPAdes (Anton et al. Citation2012), (ii) filtering plastid-like contigs, (iii) visualizing and editing de novo assembly graph using Bandage (Ryan et al. Citation2015), and (iv) checking and adjusting the annotations using Geneious 11.0.3 (http://www.geneious.com, Kearse et al. Citation2012). The tRNA genes were annotated with ARAGORN (Laslett and Canback Citation2004). For phylogenetic reconstruction, we downloaded the cp genomes of 10 species in the Hydrangeaceae family, including Hydrangea densifolia (MN380652), Decumaria barbara (MN380684), D. sinensis (MN380685), Schizophragma hydrangeoides (KY412467), Deutzia crassifolia (MG524993), D. compacta (MN380704), Philadelphus calvescens (MN380700), Kirengeshoma palmata (MN380656), Carpenteria californica (MN380687), and Whipplea modesta (MN380692). Mukdenia rossii (MG470844) from Saxifragaceae was used as an outgroup. The aligned matrix was implemented in MAFFT (Katoh and Standley Citation2013). Phylogenetic analysis was performed with maximum likelihood (ML) method, using RAxML (Stamatakis Citation2006; 2014) with 1000 bootstrap replicates.
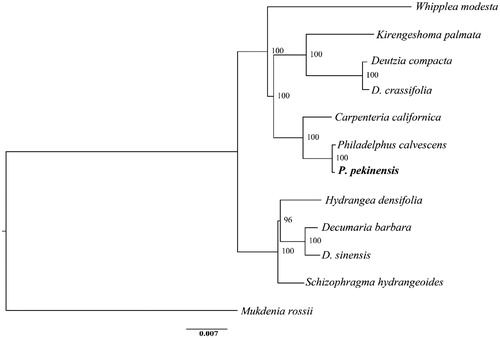
The annotated cp genomic sequences of P. pekinensis were deposited in GenBank (MN938497). The whole cp genome is 156,876 bp in length and has the typical quadripartite structure, including large single-copy (LSC) region of 86,457 bp, small single-copy (SSC) region of 18,735 bp, and two separated inverted region (IRs) of 26,058 bp. A total of 134 genes are identified consisting of 85 protein-coding genes (PCGs), 8 rRNA genes, and 37 tRNA genes. Among these genes, 61 PCGs and 22 tRNA genes are located in the LSC region (including one interregional gene rps19), while 12 PCGs and 1 tRNA gene occur in the SSC region (including one interregional gene ycf1). All the eight rRNA genes are duplicated in the IR regions. Each of the IR regions contains six PCGs and seven tRNA genes. Each of the 19 genes contains one intron, while the remaining three (ycf3, clpP, and rps12) possess two introns each. The overall GC content is 43.1%. A well supported phylogenetic tree is reconstructed, suggesting a monophyly formed by P. pekinensis and P. calvescens (). The two species demonstrate a comparably closer phylogenetic relationship with Carpenteria californica than other species.
Figure 1. Maximum likelihood tree based on 11 complete chloroplast genomes of Hydrangeaceae and one outgroup species. Numbers in the nodes are bootstrap support values based on 1000 replicates. Philadelphus pekinensis is displayed in bold.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Anton B, Sergey N, Dmitry A, Alexey AG, Mikhail D, Alexander SK, Valery ML, Sergey IN, Son P, Andrey DP, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19:455–477.
- Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemic Bullet. 19:11–15.
- JianJun J, WenBin Y, JunBo Y, Yu S, TingShuang Y, De-Zhu L. 2018. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. bioRxiv. 256479.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28(12):1647–1649.
- Laslett D, Canback B. 2004. ARAGORN, a program to the detection of transfer RNA and transfer-messenger RNA genes in nucleotide sequences. Nucleic Acids Res. 32(1):11–16.
- Ryan RW, Mark BS, Justin Z, Kathryn EH. 2015. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics. 31:3350–3352.
- Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 22(21):2688–2690.
- Stamatakis A. 2014. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.