
Abstract
Catalpa ovata G. Don is a precious Catalpa tree known as a source of valuable timber and compounds for medicinal usage. In this study, we reported its complete chloroplast genome assembled from Illumina pair-end sequencing data. The completed plastome was 158,355 bp in length, including a large single copy (LSC) region of 85,007 bp, a small single copy (SSC) region of 12,676 bp, and a pair of inverted repeat (IR) regions of 30,336 bp. The overall GC content of the genome was 38.07%, and the corresponding values of the LSC, SSC and IR regions were 36.42%, 33.54%, and 41.33%, respectively. A total of 129 genes were annotated, including 83 protein-coding genes, 38 tRNA genes, and 8 rRNA genes. Phylogenetic analysis strongly supported that Tecomaria capensis was the most closely related species to C. ovata. The newly characterized complete chloroplast genome of C. ovata will provide essential data for further investigation of this precious species.
Keywords:
Catalpa ovata G. Don, belonging to the genus Catalpa (Bignoniaceae), is a precious deciduous broad-leaved tree species producing valuable timber and compounds for medicinal usage (Kil et al. Citation2017). It is distributed to the Yangtze river basin and its north of China, as well as Korea and Japan (Park et al. Citation2010). Despite the important research value, fundamental genetic information about C. ovata remains less. In this study, we reported the complete chloroplast genome sequence of C. ovata based on Illumina pair-end sequencing data, which will provide informative data both for the conservation of this species and for the phylogeny of genus Catalpa.
Leaves of C. ovata were collected from the western campus of Hebei Agricultural University, Hebei Province (38°49′29″N, 115°26′34″E). The specimen was stored at Key Laboratory of Tree Breeding and Cultivation of National Forestry and Grassland Administration (specimen code Zishuhb1901). The total genomic DNA was extracted using modified CTAB protocol (Wang et al. Citation2015) and sequenced using the Illumina Hiseq X Ten (Illumina, CA, USA). The raw reads were filtered using Cutadapt version 1.9.1 (Martin Citation2011). Clean reads were first aligned to Tecomaria capensis (NC_037462) and then assembled using Velvet v1.2.10 (Zerbino and Birney Citation2008) and NOVOPlasty v2.7.2 (Dierckxsens et al. Citation2016). The genome was annotated using GeSeq (Tillich et al. Citation2017) and annotation for tRNA was complemented with tRNAscan-SE v2.0 (Lowe and Chan Citation2016). The annotated genomic sequence was then deposited into GenBank with accession number MT186670.
The chloroplast genome of C. ovata was a quadripartite circular with 158,355 bp, which comprised a large single copy (LSC) region of 85,007 bp and a small single copy (SSC) region of 12,676 bp, separated by 2 inverted repeat (IR) regions of 30,336 bp, respectively. This chloroplast genome encoded 129 genes, including 83 protein-coding genes, 38 tRNA genes and 8 rRNA genes. The GC content of the total genome was 38.07%, whereas the IR region had a higher GC content (41.33%) than LSC (36.42%) and SSC (33.54%).
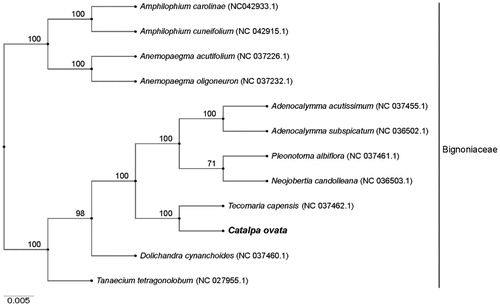
To further identify the phylogenetic position of C. ovata, 11 published chloroplast genomes of Bignoniaceae (Adenocalymma acutissimum, A. subspicatum, Amphilophium carolinae, A. cuneifolium, Anemopaegma acutifolium, A. oligoneuron, Dolichandra cynanchoides, Neojobertia candolleana, Pleonotoma albiflora, Tanaecium tetragonolobum, and T. capensis) were obtained from NCBI and aligned using MAFFT v7.313 (Katoh and Standley Citation2013). The Neighbor-Joining (NJ) tree was constructed with RAxML v8.2.11 (Stamatakis Citation2014) and the branch support was estimated with 1000 bootstrap replications (). The results revealed that T. capensis was the most closely related species to C. ovata with 100% bootstrap support.
Figure 1. Phylogenetic tree inferred by the Neighbor-Joining (NJ) method from 12 complete chloroplast genomes. All the sequences were downloaded from NCBI GenBank.
Acknowledgements
We thank Shu-gang Zhao for collecting the leaf samples.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Dierckxsens N, Mardulyn P, Smits G. 2016. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45(4):e18–e18.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Kil YS, Kim SM, Kang U, Chung HY, Seo EK. 2017. Peroxynitrite-scavenging glycosides from the stem bark of Catalpa ovata. J Nat Prod. 80(8):2240–2251.
- Lowe TM, Chan PP. 2016. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44(W1):W54–W57.
- Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17(1):10.
- Park BM, Hong SS, Lee C, Lee MS, Kang SJ, Shin YS, Jung J-K, Hong JT, Kim Y, Lee MK, et al. 2010. Naphthoquinones from Catalpa ovata and their inhibitory effects on the production of nitric oxide. Arch Pharm Res. 33(3):381–385.
- Stamatakis A. 2014. Raxml version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.
- Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, Greiner S. 2017. GeSeq – versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45(W1):W6–W11.
- Wang H, Pan G, Ma Q, Zhang J, Pei D. 2015. The genetic diversity and introgression of Juglans regia and J. sigillata in Tibet as revealed by SSR markers. Tree Genet Genomes. 11(1):804.
- Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18(5):821–829.